
More Information
Submitted: 27 June 2019 | Approved: 12 July 2019 | Published: 15 July 2019
How to cite this article: Al-Anazi KA, Al-Anazi WK, Al-Jasser AM. The beneficial effects of varicella zoster virus. J Hematol Clin Res. 2019; 3: 016-049.
DOI: 10.29328/journal.jhcr.1001010
Copyright License: © 2019 Al-Anazi KA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Varicella zoster virus; Vaccination; Bone marrow microenvironment; Hematopoiesis; Mesenchymal stem cells; Dendritic cells; Open reading frames; Exosomes; Cytokines; Signaling pathways

The beneficial effects of varicella zoster virus
Khalid Ahmed Al-Anazi1*, Al-Anazi WK2 and Al-Jasser AM3
1Department of Hematology and Hematopoietic Stem Cell Transplantation, Oncology Center, King Fahad Specialist Hospital, Saudi Arabia
2Section of Cytogenetics, Department of Pathology, King Fahad Specialist Hospital, Saudi Arabia
3Department of Research and Studies, General Directorate of Health Affairs, Riyadh Region, Ministry of Health, Riyadh, Saudi Arabia
*Address for Correspondence: Khalid Ahmed Al-Anazi, Consultant Hemato-Oncologist and Chairman, Department of Hematology and Hematopoietic Stem Cell Transplantation, Oncology Center, King Fahad Specialist Hospital, P.Box: 15215, Dammam 31444, Saudi Arabia, Tel: 966-03-8431111; Fax: 966-13-8427420; Email: [email protected]
Varicella zoster virus behaves differently from other herpes viruses as it differs from them in many aspects. Recently, there has been growing evidence on the beneficial effects of the virus in immune compromised hosts and these effects are translated into prolongation of survival. The reported beneficial effects of the virus include: (1) stimulation of bone marrow activity in patients with hematologic malignancies and bone marrow failure syndromes, (2) antitumor effects in various hematologic malignancies and solid tumors, and (3) association with graft versus host disease which has anticancer effects. Additionally, there are several reports on the safety of the live-attenuated even in severely immune suppressed individuals and on the emerging role of the virus in cancer immunotherapy. In this review, the following aspects of the virus will be thoroughly discussed: (1) new data on the genetic background, pathogenesis, vaccination, and new therapeutic modalities; (2) bone marrow microenvironment and hematopoiesis; (3) cells involved in the pathogenesis of the virus such as: mesenchymal stem cells, dendritic cells, natural killer cells, T-cells and mononuclear cells; (4) cellular proteins such as open reading frames, glycoproteins, promyelocytic leukemia protein, chaperons, and SUMOs; (5) extracellular vesicles, exosomes, and micro-RNAs; and (6) signaling pathways, cytokines, and interferons.
The reported beneficial effects of varicella zoster virus
The positive effects of the virus on marrow function and malignancies: In a single center, retrospective case-controlled study that included 16 episodes of varicella zoster virus (VZV) infection occurring in 14 patients with various types of hematologic malignancies (HMs) and bone marrow (BM) failure syndromes subjected to various forms of immunosuppressive therapies, cytotoxic chemotherapy and hematopoietic stem cell transplantation (HSCT), Al-Anazi KA, et al. reported an increase in the 3 components of blood [white blood cell (WBC) count, hemoglobin (Hb) level, and platelet (PLT) count] starting approximately 6 weeks following VZV infection [1]. This stimulation of the 3 hematopoietic cell lines in the BM that followed VZV infection was maintained for periods longer than 3 years post-VZV infection. The study clearly showed that VZV behaves differently from other members of the herpes group of viruses such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV) and that VZV can cause stimulation of BM activity starting 6 weeks after VZV infection and lasting for several years thereafter [1]. Al-Anazi KA, et al. postulated that immunological changes induced by VZV infection particularly cytokine release could be responsible for the stimulation of BM activity by VZV infection [1]. In another single center retrospective study that included 191 patients with multiple myeloma (MM) treated initially with cytotoxic chemotherapy, bortezomib-based or thalidomide-based therapy then subjected to high-dose melphalan followed by autologous HSCT, Kamber C, et al. reported that approximately 30% of these patients developed VZV infections either before or after (HSCT) [2]. VZV infections were encountered more frequently in patients with advanced stage of the disease, renal failure and relapsing MM [2]. Despite encountering VZV infections in patients with worse expected prognosis, the overall survival (OS) in patients who developed VZV infection was superior to that in patients who never developed the infection. Additionally, there was no delay in neutrophil recovery post-HSCT in patients infected with VZV and PLT count recovery post-HSCT occurred earlier in patients infected with VZV [2].
Recently, Al-Anazi KA, et al. reported reversal of pure red cell aplasia (PRCA) by VZV infection [3]. A patient with BM biopsy proven PRCA was initially treated with immunosuppressive therapy, but this treatment was discontinued due to intolerance reported by the patient. Two months after stopping cyclosporine-A and prednisolone, the patient developed localized herpes zoster (HZ) infection that was successfully treated with valaciclovir [3]. Six weeks after the VZV infection, Hb level started to increase gradually till the patient became packed red blood cell transfusion independent few months later. The steady increase in Hb level continued till it plateaued about 14 months after VZV infection. A repeat BM biopsy showed resolution of the severe erythroid hypoplasia and regeneration of the erythroid precursors in the BM [3]. Interestingly, this report confirmed not only the time line for VZV to start its effect on the 3 cell lines in the BM, but also confirmed that VZV infection may cause stimulation of BM function in patients with HMs and BM failure which may last for years as reported by Al-Anazi KA, et al. in their retrospective study published in 2005 [1,3].
Graft versus host disease and its association with VZV: Acute and mild chronic graft versus host disease (GVHD) are associated with brisk recovery of humeral immunity after HSCT while moderate to severe degrees of GVHD are associated with impaired immunological recovery following HSCT. Immune suppression is a major contributor to viral infections or reactivations of these infections following HSCT [4]. Immunosuppressive therapies, including corticosteroids, given to control GVHD are associated with increased risk of infectious complications [5]. Bacterial and viral infections can theoretically contribute to the elevation of inflammatory cytokines after allogeneic HSCT, ultimately leading to aggravation of acute GVHD [6,7]. The following strategies have been used to enhance graft versus cancer (GVC) effects: (1) tapering then discontinuation of immunosuppressive therapies in recipients of allogeneic HSCT not encountering GVHD; (2) use of donor lymphocyte infusions; (3) infusion of genetically manipulated cells or selected cellular populations such as: dendritic cells (DCs), CD8+ memory T-cells, and chimeric antigen receptor (CAR)-mediated T-cells; (4) oncolytic viruses such as myxoma virus to control GVHD while preserving GVC effect; and (5) mesenchymal stem cells (MSCs) that can used not only in the prevention but also in the treatment of GVHD have been shown to exert GVC effect while prolonging OS following allogeneic HSCT [8-18]. Interestingly, several studies have demonstrated that VZV infection may trigger chronic GVHD following allogeneic HSCT [19-21]. GVHD is associated with GVC effects and provided GVHD is of low-grade, it can be associated with improvement in OS in patients with acute leukemia or lymphoma [22-24].
Oncolytic viruses and the rising role of VZV: Viruses have 2 opposing faces: on one hand they can induce harm and disease with early as well as late complications that may be associated with significant morbidity and mortality and rarely cellular transformation and cancer; while on the other hand, viruses may provide hope to effectively treat several serious medical illnesses [25,26]. Examples of the usefulness of certain viruses in the treatment of specific diseases include: (1) use of viruses as vaccines, (2) use of genetically engineered or naturally occurring viruses as anticancer agents in the setting of oncolytic virus therapy, and (3) use of viruses as vectors in: induced pluripotent stem cells (iPSCs), gene therapy for various hereditary and acquired diseases, as well as CAR T-cell therapy [25-33]. Oncolytic viruses preferentially or selectively replicate in and subsequently kill cancer cells and they spread within the tumor without causing damage to surrounding healthy or normal tissue [34-36]. Oncolytic viruses can be used in combination with cytotoxic chemotherapy to have synergistic anticancer effects as they can efficiently kill cancer stem cells (CSCs) in several cancers [31,33,34].
Alpha-herpes viruses can induce apoptosis, autophagy and necrosis through different molecular mechanisms. These pathways influence infection and replication of alpha-herpes viruses and therefore they may become additional candidates for cancer therapy [37]. As an efficient oncolytic virus, herpes simplex virus (HSV) has the following advantages: (1) quick replication in cells and infection of multiple types of cancer cells, (2) easy modification and insertion of its large genome, (3) prevention by antiviral agents, (4) modification of its glycoprotein can improve targeting of tumor cells, and (5) ability to escape the immune response of the host in order to: (a) complement and incapacitate immune globulins via viral glycoproteins, (b) block maturation of antigen presenting cells, (c) inhibit production of cytokines and chemokines from infected cells, (d) evade the host immunological surveillance, and (e) inhibit cell death and apoptosis induced by cytotoxic T-lymphocytes [35]. So far, VZV is the only virus consistently reported to have an inverse association with glioma suggesting a protective effect of VZV against glioma [38-40]. Studies have shown the following: (1) the protective effect of VZV against the tumor is stronger for high-grade glioma, (2) the protective effect of prior VZV infection against the incidence of glioma may be mediated by the VZV-specific T-lymphocytes, (3) VZV exhibits an intrinsic oncolytic potential in malignant glioma cultures and might become a novel candidate for virotherapy in glioblastoma multiforme, and (4) human MSCs are suitable for delivering VZV to the sites of tumor growth [38,39,41]. However, efficacy of oncolytic virotherapy in malignant glioma has the following difficulties: (1) poor penetration of the viral particles across the blood brain barrier (BBB), (2) ineffective transduction of sufficient numbers of malignant glioma cells, (3) poor oncolytic potential, (4) limited tumor cell selectivity of stable gene delivery, and (5) uncontrolled host immune reaction with considerable adverse effects and complications of the viral infections [41].
Update on VZV infections and their management
The virus and its genome: VZV is a human neurotropic virus which is highly contagious. It is an exclusively human pathogen and this makes is extremely difficult to find an animal model for the virus [42-44]. VZV is a double stranded DNA virus that belongs to the alpha group of herpes viruses. VZV genome is approximately 125 kbp in size and is the smallest among herpes viruses. VZV genome, which has 74 open reading frame (ORF) proteins, consists of a linear double-stranded DNA molecule [45-48]. The genome consists of 2 main coding areas: the unique long segment and the unique short segment, each of which is flanked by internal repeat and terminal repeat sequences [46-48]. The virion is composed of an icosahedral nucleocaspid; that harbors the DNA genome; surrounded by a tegument layer which is covered by an envelope derived from the host cell or a plasma membrane with incorporated viral glycoproteins as shown in figure 1 [45-48]. Over approximately 70 million years of evolution, the VZV genome has lost almost all the genes that are not essential for its survival [47]. Relatively small genomes and high proliferation rates allow viruses to accumulate mutations and continuously present the host with new challenges. Consequently, viruses either escape detection or modulate host physiology often by redirecting cellular pathways to their own advantage [49].
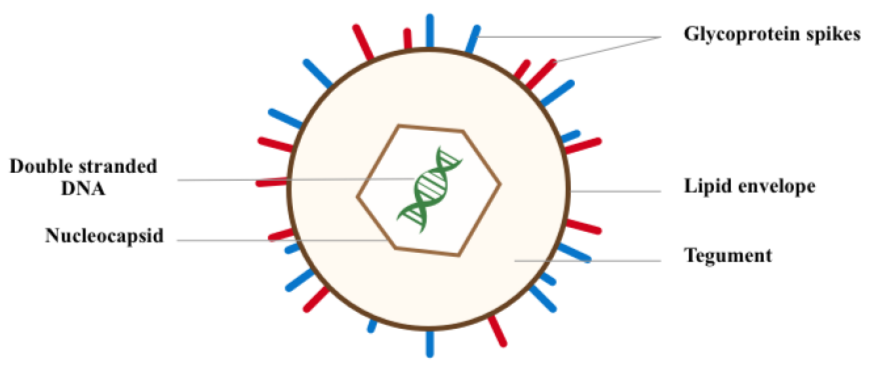
Figure 1: Showing the varicella zoster virus genome.
Epidemiology and risk factors: There are several risk factors for the developments of VZV infections. These predisposing factors are shown in table 1 [3,50-78]. Initial studies have identified the following 4 geographical VZV genotypes: genotype A in Africa and Asia, genotypes B and C in North America and Europe, and Genotype J in Japan and South Korea [79,80]. Single-nucleotide polymorphism and restriction fragment length polymorphism are used in detecting these genotypes [79-84]. Recently, the following new genotypes: E1, E2, M1, M2, M3, M4, VI, VII, VIII, IX have been described in: Germany, Czech Republic, Spain, France, Australia, and New Zealand [81-88].

Table 1: Risk factors for varicella zoster virus infections.
New data on the pathogenesis of VZV infections:
VZV pathogenesis: Primary VZV infection causes viremia in T-lymphocytes. Viremia induces the characteristic skin rash [89,90]. Subsequently the virus migrates in a retrograde manner via sensory neurons into dorsal root ganglia where latency is established. Later on, VZV reactivation from dorsal nerve ganglia causes antegrade travel of the virus to cause the dermatomal disease of HZ [89-91]. VZV establishes latency in multiple cranial and dorsal root ganglia as well as thoracic sympathetic and enteric ganglia [90,92]. VZV-infected lymphocytes are used to induce latent infection in sensory and enteric neurons. However, evidence suggests that exosomes and stimulator of interferon (IFN) genes may play roles in the establishment of neural latency by preventing proliferation [90,93]. During productive infection, the complete proteome of VZV is expressed through the interaction between a small number of viral transcriptional activators and the general transcription apparatus of the host cell [94]. Productively infected cells frequently form multinucleate syncytia consisting of fused cells. These syncytia are present in human skin, ganglionic tissue, and tissue cultures [95]. The interaction between VZV-immediate early (IE) 63 protein with human antisilencing function 1 protein may help to regulate transcription of viral or cellular genes during lytic and/or latent infection [96,97]. The cellular component host cell factor-1 is a key factor for controlling VZV-IE gene expression by functioning as the common element for distinct factors cooperating at the IE gene enhancers [98]. The major viral transactivator, commonly designated the IE-62 protein, interacts with the human mediator of transcription [94].
Pathogens have evolved strategies to promote their survival by dramatically modifying the transcriptional profile and protein content of the host cells they infect. Thus, pathogens are able to induce long-term and heritable changes that are essential to the pathogenesis of infectious diseases and persistence of pathogens within the hosts [97]. VZV is a highly fusogenic virus. Fusion of VZV-infected cells is a consequence of virally expressed glycoproteins. Fusion permits entry of VZ virion at the plasma membrane and into the intracellular cytoplasm [95]. Herpes viruses possess complicated mechanisms to seize various host cellular components for immune evasion, replication, and virion egress [99]. Viruses have evolved in tight association to the host cell to be able to hijack the cellular apparatus that is necessary for their replication. They depend on many aspects on the cellular machinery of the host in order to replicate efficiently [100]. Exosomes can shuttle various molecular cargo from a donor to a recipient cell. They serve as important vehicles facilitating cell-to-cell communication [99]. Cell-to-cell fusion induced by VZV infection has long been known to occur among fibroblasts and keratinocytes during formation of vesicles in the skin in both primary (chickenpox) and secondary (HZ) infections [90,91,95,101].
The virus-host interactions: Viruses routinely manipulate the host cell cycle to create a favorable environment for replication and while evading detection, viruses utilize a diverse array of strategies and molecular targets to subvert cellular processes and these include: (1) cell-to-cell regulation, (2) major histocompatibility complex (MHC)-restricted antigen presentation, (3) intracellular protein transport, (4) apoptosis, (5) cytokine-mediated signaling, and (6) hormonal immune responses [49]. Virus infection of mammalian cells activates DNA damage response pathways to enhance viral replication. Also, cells respond to DNA damage by activating checkpoint pathways that: delay the progression through the cell cycle, promote DNA repair, and induce cell repair [102]. In classical human infections, VZV rarely infects dividing cells such as skin fibroblasts, differentiated keratinocytes, mature T-cells, and neurons none of which are actively synthesizing DNA [103]. However, VZV is able to productively infect these cells and use their machinery to replicate the viral genome. VZV infection of human foreskin fibroblast cells results in atypical cyclin expression and cyclin-dependent kinase activity [103].
Transcription of the virus is strictly regulated by cascade-like processes: expression of IE transcripts, and expression of early then late kinetic classes of transcripts. The viral E genes encode proteins that are used in DNA replication, while viral L genes code for the structural elements of the virus [104]. While multiple alternatively spliced VZV latency associated transcript (VLT) - isoforms are expressed during lytic infection, a single unique VLT isoform; which specifically suppresses ORF 61 gene expression in co-transfected cells; predominates in latently VZV infected human trigeminal ganglia [104]. Activation of: (1) H2A which is a member of histone family that interacts with eukaryotic DNA and helps to regulate transcription; and (2) ATM (ataxia telangiectasia-mutated) in VZV-infected cells is associated with the expression of specific VZV genes [102]. Both VZV-ORF 28 and VZV-ORF 29 genes, which can be expressed either coordinately or independently, are expressed during VZV lytic infection but only the latter is expressed in latently infected neurons. However, the observed expression of only VZV-ORF 29 gene during VZV latency may involve neuron specific cellular factors and/or structural aspects to the latent viral genome [105]. The virions could deliver proactive cellular kinase to non-dividing cells that normally do not express it. Cellular kinases play an important role in the phosphorylation of VZV proteins [106]. IE 62, which is a constituent of the virion tegument, is a substrate for cyclin-dependent kinase 1/cyclin B [106]. The time line for primary and secondary VZV infections with the main pathological events and associated immunological changes as well as the BM stimulatory effects are illustrated in figure 2 [1,3,89-91,95,101,106].
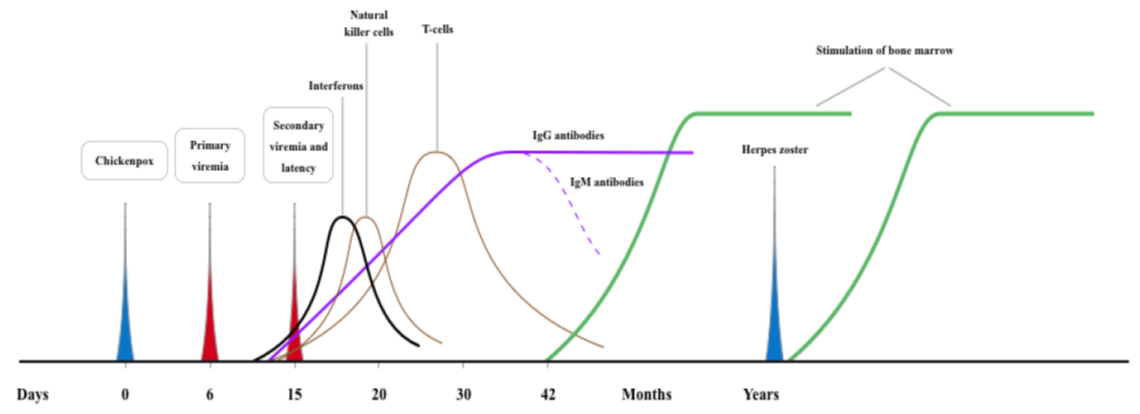
Figure 2: Timeline for pathological events and bone marrow consequences of varecella zoster virus infections.
Autophagy: Autophagy or self-eating involves degradation of cytoplasmic constituents in lysosomes which are able to break down all cellular macromolecules including lipids, polysaccharides and proteins by means of their hydrolases [107]. Autophagy is an ancient survival strategy and a well-recognized catabolic process by a stressed cell during which misfolded or damaged proteins are engulfed within double-walled cytoplasmic granules called autophagosomes [47,89,108,109]. The 3 major autophagy pathways that exist in mammals include: microautophagy, macroautophagy, and chaperone-mediated autophagy [107,110,111]. The molecular machinery of autophagy which generates autophagosomes in cells was originally described by Yoshinori Ohsumi who was awarded Nobel prize in physiology and medicine in 2016 [107]. Autophagy is a highly conserved pathway among eukaryotes that involves recognition, capture, and trafficking of various intracellular components to the lysosome for degradation. So, it is responsible for maintaining cellular homeostasis in response to various internal and external stimuli [110,111]. Autophagy is tightly controlled by a set of autophagy-related gene proteins and secretory autophagy is a mechanism by which viruses replicate and spread [110].
The discovery of lysosomes by Christian de Duve in 1955 was a landmark in studying intracellular protein degradation [110]. Herpes viruses recruit autophagic membranes into their envelopes [107]. Autophagy is closely associated with VZV infection [47]. Unlike HSV, VZV genome has no inhibitors of autophagy [47]. Autophagy within a VZV-infected cell is remarkably different from autophagy within a HSV-infected cell [108]. VZV-induced autophagy facilitates VZV glycoprotein biosynthesis and processing [91]. During VZV infection autophagy is up-regulated and autophagic flux is increased, while inhibition of autophagy leads to a marked reduction in viral spread. Thus, VZV partially inhibits the late-stage of mTOR-mediated autophagic flux and the inhibitory effects are more pronounced when the cells are under stress [109]. Also, inhibition or block of autophagic flux may yield higher VZV titers [111].
The role of epigenetics in herpes viruses and VZV: Modulation of protein acetylation via histone deacetylases (HDACs) is a critical regulatory factor during herpes virus infection [112]. Viruses have evolved a wide array of mechanisms to destroy HDAC functions [112,113]. Viral genomes need chromatin for protection and execution of their gene expression programs. The assembly and distribution of active (euchromatic) chromatin or repressive or heterochramatin on viral genome can determine the fate of the virus including the ability to establish latent infection [114-116]. Most viruses struggle to modulate and utilize the chromatin machinery of host cells to promote efficient lytic infection and to control persistent latent states [117]. Telomeres and viruses utilize common mechanisms to maintain genome integrity and regulate innate immunity. Telomeres may provide host cells with antiviral functions by trapping invading genomes and suppressing their expression through inaccessible heterochromatic structures [118]. Epigenetic manipulation using DNA methyltransferase inhibitors and HDAC inhibitors may potentially become novel epigenetic antiviral therapies [119]. Inhibition of the histone demethylase LSD1 results in accumulation of repressive chromatin and blockade of viral genome expression and ultimately inhibition of α-herpes virus lytic as well as latent infections [115].
Clinical manifestations and complications of VZV infections
Primary infection usually occurs in childhood and causes chickenpox. After primary infection, VZV becomes latent in nerve ganglia (dorsal root, cranial nerve, trigeminal, and autonomic ganglia) [42,58]. Reactivation may occur decades later and the virus causes HZ infection with typical painful skin rash that has characteristic dermatomal distribution [42,58]. The predisposing factors for reactivation of VZV infection are shown in table 2 [42,58,120,121]. In severely immune compromised individuals, pre-existing antibody does not prevent VZV reactivation, but may contribute to decreased viral load thus resulting in mild clinical course [120]. The clinical manifestations and complications of VZV infections are shown in table 3 [42,58,121-127]. In immune compromised hosts having VZV infection, atypical skin eruption may be encountered and disseminated infection may also develop even in the absence of skin lesions [58,120].

Table 2: Predisposing factors for reactivation of varicella zoster virus infections.

Table 3: Clinical manifestations and complications of varicella zoster virus infections.
Laboratory diagnosis of VZV infections
The diagnosis of VZV infection is usually made on clinical grounds based on the presence of the characteristic skin lesions of chickenpox or HZ [42,90,128]. However, additional diagnostic techniques may be needed to confirm the diagnosis and these include: (1) virus isolation by culture which carries a low yield rate, (2) serology using enzyme-linked immunosorbent assay (ELISA), (3) direct fluorescent antibodies on scrapings obtained from active skin lesions, and (4) real-time polymerase chain reaction (RT-PCR) which has higher sensitivity than serological assays [42,90,128]. Acyclovir resistance of VZV infections has been reported on rare occasions in immune compromised individuals such as acquired immunodeficiency syndrome (AIDS) patients and recipients of solid organ transplantation (SOT) [129-131]. The characterization of drug resistance using genetic testing of the thymidine kinase and polymerase genes has been considered the method of choice for the determination of VZV resistance to antiviral agents [129-131]. Ultra-deep sequencing, after initial detection of drug resistant mutations by Sagner sequencing, can be used in immune compromised hosts [132].
Treatment, vaccination and antiviral prophylaxis
Treatment of VZV infections: The available therapies for VZV infections include: (1) acyclovir which has been the standard of care for many years; (2) valaciclovir; (3) famciclovir; (4) bromovinyl deoxyuridine (brivudine); and (5) bicyclic pyrimidine nucleotide analogues (BCNAs) [1,133-136]. In immune compromised individuals, high-dose acyclovir is usually recommended intravenously for a total duration of 7 to 10 days [1,3,135,136]. Brincidofovir has been successfully used in the treatment of acyclovir resistant disseminated VZV infection in immune compromised hosts such as recipients of HSCT having GVHD [137]. Also, intravenous and intravitreal foscarnet have been successfully used in the treatment of acyclovir resistant acute retinal necrosis (ARN) caused by VZV infections [138-140]. BCNAs have been found to be more potent against clinical isolates of VZV than acyclovir or brivudine. However, BCNAs are not active against VZV strains that are resistant to acyclovir or brivudine and that bear mutations in the viral thymidine kinase gene [134]. A novel anti-VZV compound (35 B2 derivative of pyrazolo-1,3,5-triazin-4-one) can inhibit both acyclovir-resistant and acyclovir-sensitive strains of VZV by targeting herpes virus major capsid protein and inhibiting normal capsid formation [141]. Other new therapeutic agents for the treatment of VZV infections include: (1) aryl bicyclic nucleoside analogues such as FV-100; (2) BCNAs as various types of these agents have been found to be promising future therapies for VZV infections; and (3) bicyclic aryl furano pyrimidines [133,142,146]. For post-herpetic neuralgia (PHN), gabapentin as well as local and systemic analgesics are usually prescribed [135,147,148].
VZV vaccines: There are two types of VZV vaccines: (1) varicella vaccines such as varilrix- in the United Kingdom, varivax-in the United States of America (USA), and the combined measles, mumps and rubella and varicella vaccine, all of which contain live-attenuated oka strain of VZV; and (2) HZ vaccines that include zostavax, and HZ/su [91,149,150]. Zostavax is the only HZ vaccine that is currently approved in Europe and the USA. It contains the live attenuated VZV oka stain and it is given as one injection subcutaneously. It is recommended in immunocopetent adult’s ≥ 60 years old with overall efficacy of 51.3%. It reduces the incidence of HZ by 51% within a 3 year period but a significant reduction in vaccine-induced immunity is observed within the first year after vaccination [91,149,150]. HZ/su is a subunit vaccine candidate that has recently shown improved efficacy of HZ prevention in 2 phase III clinical trials. It is non-live, recombinant subunit glycoprotein E+adjuvant ASO1. It is given intramuscularly twice. It is recommended for immune competent individual’s ≥50 years with overall efficacy of 97.2% [91,149,150]. Post exposure immune globulins or immunoglobulin prophylaxis with ZariZIG is usually given to individuals having recent contact with patients infected with VZV [149,151]. The main indications of VZV vaccination include: health care providers, post exposure prophylaxis, and individuals ≥50 years of age [43,151,152]. However, VZV vaccination is traditionally contraindicated in the following groups of patients: (1) patients having solid tumors or HMs particularly those on cytotoxic chemotherapy or novel agents; (2) recipients of SOT or HSCT receiving immunosuppressive therapies; (3) patients having autoimmune and collagen vascular disorders treated monoclonal antibodies; (4) AIDS patients; (5) patients receiving long-term high dose corticosteroids; and (6) patients having active VZV infections [43,151,152].
Before the development of VZV vaccines, there was almost universal infection with VZV which has become endemic worldwide [153]. Developments in varicella and HZ vaccines which require a better understanding of the host response to VZV can offer the potential to prevent the majority of VZV infections [150,153]. The implementation of universal varicella vaccination in many areas around the globe >20 years ago has resulted in a significant reduction in the burden of varicella-associated disease [154]. Both VZV infection and varicella vaccination can induce VZV-specific antibodies and T-cell mediated immunity that are essential for recovery [154]. Several countries have also adopted VZV vaccination for high-risk groups [154,155]. Despite the rare reports of breakthrough VZV infections that may become disseminated and life-threatening particularly in immune compromised hosts, VZV vaccines including the live-attenuated ones are generally safe as shown by a 10 year global safety database as well as 2 systematic literature reviews, each of which included at least 31 million doses of VZV vaccines administered [151,156-159]. VZV vaccines including the live attenuated ones have been shown to be safe in recipients of: SOT as well as HSCT, both autologous and allogeneic, in addition to patients with HMs and solid tumors [160-168]. Additionally, VZV vaccines have been shown to be effective and safe in: (1) patients with diabetes mellitus, autoimmune disorders and renal disease, (2) elderly individuals on corticosteroid maintenance therapy and those living in long-term care facilities, and (3) individuals with history of HZ infection [169-173].
Prophylaxis against reactivation of VZV infections: Reactivation of VZV infections may occur in patients with various HMs and in recipients of autologous as well as allogeneic HSCT [77,174-177]. Reactivation of VZV infections in these severely immune compromised individuals may be associated with complications such as disseminated infections, significant morbidity, and mortality rates that may reach 34% [77,174-179]. Therefore, prevention of reactivation of VZV infections in this group of patients; particularly those with MM, low lymphocytic count, and those on long-term corticosteroid therapy; is needed to prevent complications of VZV infections [77,174-177,179,181]. Acyclovir prophylaxis is recommended in patients with: (1) HMs receiving intensive chemotherapy or novel agents, and (2) recipients of autologous and allogeneic HSCT [174-177, 179,181,182]. Initially, there was a trend to give acyclovir prophylaxis for up to 6 or 12 months in recipients of autologous and allogeneic HSCT respectively [174,178,180,182]. Nowadays, the recent literature is in favor of giving antiviral prophylaxis for longer than one year in recipients of HSCT [174,175,177,178,180,182]. Several retrospective studies have shown that extended prophylaxis with acyclovir has been shown to be safe and effective [175,180]. However, the benefits and safety of long-term prophylaxis with low-dose acyclovir should be confirmed in large prospective trials as long-term use of acyclovir may be associated with not only side effects, but also with evolution of drug resistance [180,182].
Animal and other experimental models for VZV
VZV pathogenesis, latency, and reactivation are difficult to study due to the fact that VZV is an exclusively human pathogen [150,183,184]. Due to the strict host specificity of infection and cell-associated nature of the virus, our knowledge of host-pathogen interaction regarding VZV infection and VZV pathogenesis, latency and reactivation remains incomplete [150,183,185]. Development of more efficacious second generation vaccines and antiviral therapies against VZV as well as better understanding of the host response to VZV infection are hampered by the scarcity of animal models that recapitulate all aspects of VZV infection including virological, immunological and pathological hallmarks of both acute and latent VZV infection in humans [150,183-186]. Normal human neuronal progenitor cells in tissue-like assemblies have been shown to be an effective system to investigate the long-term interactions between VZV and the complex assemblies of human neuron cells [183]. Experimental inoculation of mice and other non-human primates (NHPs) with VZV has produced seroconversion but not varicella, while simian varicella virus (SVV) has produced a naturally occurring exanthematous disease that mimics human varicella in NHPs [185,187]. Experimental SVV inoculation into rhesus macaques via intrabronchial route can reproduce the hallmarks of acute VZV infection in humans including: viremia, generalized varicella T and B cell responses, resolution of viremia and varicella, and establishment of latency in only ganglionic neurons. However, reactivation of VZV has not been experimentally induced by the rhesus macaque model [150,185]. The severe combined immunodeficiency-humanized mouse model has been applied to study VZV pathogenesis where it has facilitated rigorous evaluation of the role of several genes in vivo and it has demonstrated the following: (1) ORF-47 and ORF-66 are required for VZV replication in human T-cells, (2) ORF-47 and ORF-14 are necessary for infection and replication in skin cells, and (3) the C terminus of VZV glycoprotein M contains trafficking mofits to maintain skin virulence in studying the pathogenesis of VZV [150,188,189].
Recently, terminally differentiated neurons have received increased attention as a means to study the interactions between VZV and human neurons, but the short life-span of these cells in culture has limited their application [183]. Sensory neurons are the only types of cells that support the entire VZV life cycle. After generation of human iPSCs from skin fibroblasts, sensory neurons have been produced from these human iPSCs. Hence, generation of iPSCs and sensory neurons may advance our knowledge regarding the pathogenesis of VZV and these cells may serve as a platform for the development of new therapeutic interventions [190]. Neurons have also been generated from human embryonic stem cells (ESCs) and infection of these neurons has demonstrated axonal infection, transport of VZV, and evidence of productive neuronal infection [191]. Several studies have shown that neurons derived from human ESCs are highly permissive to a productive and spreading VZV infection and that whether the infection is productive or not depends on the infectious viral dose. Thus, these neurons may provide an experimental or in vitro model of latency and reactivation of VZV [192-195]. Over the last few years, the field of VZV latency and reactivation has greatly advanced due to the derivation of human neurons to perform mechanistic studies in vitro, and the advanced molecular techniques which have led to the identification of VLT in vivo [196]. Studies have shown that types I and II IFNs inhibit viral replication and that IFN-γ inhibits transcription, replication, and production of neurons derived from iPSCs [196-198]. Hence, IFNs could be used to induce a VZV latent phenotype in human neurons or to inhibit VZV reactivation [196]. Concentration of VZV-containing supernatant, use of debris fraction, and generation of reporter cell line are useful techniques to study virus entry and latency, detect virus, and design new antiviral agents [199-201]. Numerous efforts have been made to develop adequate animal models of VZV infection but these models remain limited because all aspects of VZV infection, latency and reactivation, understanding VZV pathology will not only remain difficult but also incomplete without a suitable model [150,184].
Bone marrow microenvironment and hematopoiesis
Bone marrow microenvironment: The BM microenvironment is the domicile of hematopoietic stem cells (HSCs) as well as the malignant processes that develop in the BM [202]. The best characterized BM microenvironment is the niche that regulates HSCs [203]. The BM niche or microenvironment has the following components: (1) cellular components such as MSCs, HSCs, and derivatives of MSCs such as: osteoblasts, adipocytes, endothelial cells, perivascular cells, and Schwann cells; and (2) functional components that are composed of the following growth factors and cytokines which regulate hematopoiesis: stem cell factor (SCF), transforming growth factor (TGF)-β, granulocyte-colony stimulating factor (G-CSF), integrated-1/β-catenin (Wnt) ligands, CXC-mofit-chemokine ligand (CXCL)-12, angiopoietin and thrombopoietin [204-207]. The following BM niche components share a common origin: peripheral sympathetic neurons, Schwann cells, and MSCs [207]. The interactions between the niche constituents and HSCs maintain hematopoiesis by: (1) regulating renewal, differentiation, and migration or trafficking of HSCs; and (2) integrating neural and hormonal signals from the periphery [208]. The main function of BM microenvironment is to provide signals that regulate and support the production of billions of blood cells which are necessary to maintain homeostasis [209]. BM niche regulates endogenous processes such as hematopoiesis but could also support the survival of tumors such as facilitating existence of CSCs in dormancy for decades [206-210]. The following are implicated in the maintenance of HSCs: endosteal fibroblasts, perivascular stromal cells including endothelial cells, CXCL-12, CXCL-12-abundant reticulin cells, leptin-receptor+ stromal cells, and nestin- GFP+ mesenchymal progenitors [211]. CXCL-12 plays a crucial role in maintaining HSC functions including retention in the BM, quiescence, and repopulating activity [208].
HSCs which give rise to all blood cells, including immature cells, are maintained and regulated by special microenvironment or niches in the BM cavity [212]. NOTCH signaling is crucial for HSC maintenance. Adipocytes are a BM niche component that promotes hematopoietic regeneration [204]. Distinct stromal or hematopoietic progenitor cells in the BM niche generate signals that regulate self-renewal, proliferation and trafficking of HSCs [211]. HSC niche supports steady-state hematopoiesis and responds to the changing needs during stress and disease [212]. Additionally, the nervous system is an important regulator of HSC niche and its influence is established early in development when stem cells are specified [212]. Neural crest-derived MSCs have regulatory pathways that control hematopoiesis in the hematopoietic niche [213]. Dyregulation between neural and hematopoietic systems can contribute to disease, thus repurposing of neuro-regulatory drugs may create new therapeutic opportunities to support hematopoiesis [212].
Steady-state and stress-induced hematopoiesis: Hematopiesis is the process by which all mature blood cells are produced [205]. New blood cells belonging to different cell lineages are produced from stem cells during embryogenesis and throughout the life-time of humans to replace cells that have completed their lifespan [214]. In hematopoiesis, which is essential for the development and survival of normal individuals, all the specialized hematopoietic cells are produced from a small number of definitive multipotent HSCs [214,215]. Hematopoiesis is a highly organized process that leads to the formation of all types of blood cells from HSCs residing in the BM, and differentiation of HSCs into immune cells through a series of lineage commitments [214,216-218]. Hematopoiesis is under tight control of a group of hematopoietic cytokines [219]. Also, hematopoiesis is a dynamic biological process that can be influenced by environmental factors such as infection or inflammation [220]. The same cytokines control basal as well as emergency hematopoietic cell proliferation [219]. However, each cytokine has multiple actions, mediated by receptors whose cytoplasmic domains contain specialized regions initiating the various responses such as survival, proliferation, differentiation commitment, maturation, and functional activation [219]. Thus, pro-inflammatory cytokines are emerging as novel and fundamental regulators of hematopoiesis. However, there are differences in the roles of some cytokines during fetal life and adulthood [217,221]. The different cytokines, chemokines, ligands, and signaling pathways that are involved in hematopoiesis for hematopoietic stem and progenitor cell (HSPC) proliferation and myeloid differentiation are shown in table 4 [215,219,221]. There are molecular mechanisms that control HSC maintenance in homeostasis and these regulatory networks orchestrate the interplay of: (1) key transcription factors such as: FOXO, PTEN, Gfi1, E47, and Notch; (2) survival genes such as: Bcl2, Mcl1, and Bcl-XL; and (3) cell cycle regulators such as: p16, p18, and p21 [214,220]. Tumor necrosis factor (TNF) is an inhibitor of hematopoietic cell proliferation, while interleukin (IL)-6 and IL-1 induce viability and differentiation without inducing cell multiplication in normal myeloid precursors [214]. IL-6 upregulates: Bcl-XL, Mcl-1, and FLIP genes and downregulates: Bcl-2 and Bax genes [214]. Regulators of multiple fates of HSCs require the cooperative actions of several cytokines and other hormones that bind to the receptors of these cells [221]. There are 7 groups of BM myeloid progenitor cells with certain transcriptional characteristics and these include: neutrophils, basophils, eosinophils, monocytes, DCs, erythrocytes, and megakaryocytes [222,223].

Table 4: The cytokines, chemokines, ligands and signaling pathways involved in hematopoiesis, myeloid differentiation and HSPC proliferation.
HSCs normally reside in specialized niches in the BM but they can circulate under stress conditions such as infection or inflammation [220,221]. HSCs repopulate the innate system during normal replenishment as well as under the burden of pathogen stress, but the respective outcomes of differentiation are not the same [220]. HSCs are maintained in a predominantly quiescent state, but they can rapidly enter cell cycle and differentiate in response to infection or inflammation [217]. During pathogen exposure, hematopoiesis may yield a progeny in proportions that are different from those produced under steady-state hematopoiesis and this may be due to: (1) pathogen engagement of Toll-like receptors (TLRs) expressed on HSCs, and/or (2) HSCs being responsive to inflammatory cytokines that are produced in response to pathogen burden and that are present in the BM microenvironment [217,220]. HSPCs respond to infection through 5 general mechanisms: (1) HSPCs respond to the depletion of downstream neutrophils, (2) they respond to the inflammatory cytokines produced by various hematopoietic and non-hematopoietic cells during infection, (3) HSPCs respond to pathogen-associated molecular patterns and danger associated molecular patterns directly through TLRs, (4) they respond to paracrine signals from the same niche, and (5) theoretically, pathogens can affect HSPC activity by infecting them [224].
In acute inflammation, types I and II IFNs, TNF, and lipopolysaccharide directly stimulate HSC proliferation and differentiation while in chronic inflammation, cytokine signaling leads to HSC exhaustion and may contribute to the development of HMs [225]. Studies have shown that cytokines and ligands which are produced during stress conditions such as infection include: (1) IFNs; (2) TNF; (3) cytokines such as; Il-1α, IL-1β, IL-3, Il-6, IL-18, IL-23, mtDNA, HMGB1, SCF, and thrombopoietin; and (4) Flt-3 ligand [224-226]. However, certain cytokines that are induced during inflammation have significant effects on HSCs in the BM [225]. Pro-inflammatory cytokines such as G-CSF indirectly affect HSCs by altering BM microenvironment, disturbing the stem cell niche, and mobilizing HSCs into the peripheral circulation [225]. HSCs respond to microbial products and inflammatory cytokines permitting alteration in hematopoiesis in response to the burden of pathogen exposure [220]. The types of BM microenvironment responses or stress-induced changes in hematopoiesis in response to infection or inflammation include: (1) enhanced output of HSPCs and the myeloid BM lineage or emergency granulopoiesis giving rise to short-lived neutrophils, basophils and eosinophils due to rapid mobilization of granulocytes and HSPCs from the BM to the peripheral tissues; (2) suppression of erythropoiesis and development of anemia or enhanced erythropoiesis as a physiological response to inflammation; (3) type I IFNs can shift HSCs from resting state or cell cycle arrest to induce proliferation and differentiation ultimately resulting in increased numbers of HSCs; (4) enhanced output of innate immune cells at the expense of lymphopoiesis and erythropoiesis; and (5) development of extramedullary hematopoiesis in the spleen and liver to open new resources for granulopoiesis and myelopoiesis so as to compensate for the diminished BM hematopoietic progenitor cells during infection [218,224]. The techniques that are used to define hematopoiesis and to trace the changes induced by various stresses such as infection include: (1) novel imaging technologies such as intravital imaging and laser-scanning cytometry; (2) conditional knockdown technologies; and (3) fluorescence-activated cell sorting [216,218].
The range of proinflammatory cytokines produced during infection or tissue injury impact the size and shape of the hematopoietic system [217]. Pathogens disturb hematopoiesis through: (1) direct effect on HSCs either by infection or through exposure to microbial products; and (2) indirect effects on the BM microenvironment that supports stem cells [220]. The effects of inflammatory signals on HSCs at baseline or resting state and during times of stress or infection are likely to depend upon the level and duration of signaling with short-term exposures facilitating the development of effective immune responses while chronic signaling may contribute to HSC dysfunction [225]. Acute microbial infection elicits profound changes in hematopoiesis with alterations in numbers and proportions of uncommitted progenitor cells. For example, sepsis is characterized by hyperactivity of the immune system manifested by excessive production of pro-inflammatory cytokines and chemokines followed by hypoactivity and neutropenia [220]. Thus, stress-induced hematopoiesis is a highly complex and dynamic process that involves crosstalk between: HSPCs, BM stromal cells, and non-hematopoietic tissues to sense the invading pathogen and to convert the signal of infection into a signal of myeloid differentiation [224]. Hematopietic failure associated with overproduction of pro-inflammatory cytokines is often a feature of: chronic inflammatory diseases, HMs, and BM failure syndromes [217]. During systemic infection, cell death can directly affect HSPCs and this ultimately leads to impaired hematopoiesis, cytopenia and immune suppression [226]. Explanation of defects in emergency hematopoiesis encountered during systemic infection include: (1) inappropriate activation of cell death, and (2) suppression of HSPC proliferation, differentiation and self-renewal. Thus, the response of the progenitor cell compartment to intracellular infection and inflammatory cytokines may be central to an effective immune response [226].
VZV-associated pancytopenia and aplastic anemia
It is well recognized that peripheral blood cytopenia is the hematological hallmark of septic shock [226]. Additionally viruses can have tremendous impact on the hematopoietic process. Examples of the consequences of viral infections on the BM include: aplastic anemia, pancytopenia, lymphoproliferative diseases, hemophagocytic lymphohistiocytosis, and a variety of other cancers [218,227,229]. Examples of the viruses that have been reported to have adverse effects on BM function are: EBV, CMV, Parvovirus B-19, human immunodeficiency virus (HIV), hepatitis A virus, hepatitis C virus, dengue virus, VZV, and HSV [1,227,229,230]. The mechanisms involved in the adverse consequences of viral infections on the BM include: (1) direct viral infection of HSPCs, (2) viral recognition of HSPCs, (3) indirect effect on HSPCs by inflammatory mediators, and (4) the role of BM microenvironment on hematopoiesis upon viral infection [227,231-234]. VZV infections have been reported to cause not only transient pancytopenia but also aplastic anemia that may require allogeneic HSCT [66,227,235-237]. Several studies have shown that HZ is associated with increased risk of developing solid tumors as well as lymphoid malignancies [238-242].
Cells involved the pathogenesis of VZV
Mesenchymal stem cells:Mesenchymal stem cells and relation to VZV: MSCs are heterogeneous, non-hematopoietic, adult multipotent stromal progenitor cells that have the capacity of self-renewal and multi-lineage differentiation [243-248]. They can be isolated from BM, peripheral blood, umbilical cord blood, amniotic fluid, and adipose tissue [243-245]. MSCs have certain distinguishing features including the characteristic surface markers [243245]. MSCs have immunomodulatory and immunosuppressive properties that enable them to have several therapeutic and clinical applications which include: (1) prevention and treatment of GVHD in recipients of allogeneic HSCT, (2) treatment of several autoimmune disorders, (3) role in regenerative medicine and tissue repair such as treatment of myocardial ischemia, cardiac dysfunction and myocardial infarction, chronic wounds, and spinal cord injuries, and (4) treatment of acute respiratory distress syndrome [243-245]. MSCs are a major constituents of HSC niche which is a highly complex and dynamic microenvironment of the BM [208]. They are defined by a set of different markers such as: nestin, neural-glial antigen-2, leptin receptors, and paired related homeo box [202]. MSCs secrete HSC-supporting factors such as: CXCL-12, angiopoetin, and SCF/kit ligand [202]. They synthesize and secrete multiple paracrine factors that are able to: affect migration of MSC, promote angiogenesis, and inhibit apoptosis [249]. Nestin+ MSCs contain all the BM colony-forming-unit fibroblastic activity and can be propagated as non-adherent mesenpheres that can self-renew and expand in serial transplantations [250]. Nestin+ MSCs are spatially associated with HSCs and adrenergic nerve fibers and they highly express HSC maintenance genes [250]. Leptin receptor is a marker that highly enriches BM-MSCs. Exosome secretome of BM-MSCs regulates stem cell maintenance and their regenerative potential [251]. The BM-derived secretome will be critical to the future development of therapeutic strategies for oncologic diseases and regenerative medicine [210]. Apparently, MSCs are the masters of survival and clonality as they communicate with diverse immune cells and interact with other cellular components of the BM microenvironment as well as with normal cells, leukemic stem cells, and progenitor cells [252].
The main functions of MSCs include: formation of hematopoietic microenvironment, modulation of the activity of the immune system, and regulating cell trafficking [253]. MSC homing is the arrest of MSCs within the vasculature of a tissue followed by transmigration across the endothelium [254]. When stimulated by specific signals, MSCs can be released from BM niche into circulation and can be recruited to the target tissues where they undergo in situ differentiation and contribute to tissue regeneration and homeostasis [255]. The efficacy of MSCs is linked to their immune suppressive and anti-inflammatory properties primarily due to the release of soluble factors [256]. Putative roles of BM-MSCs during infection: (1) phase 1, detection of pathogen and damage signal; (2) phase 2, activation of host immune response; (3) phase 3, elimination of pathogens; (4) phase 4, induction of proinflammatory gradients; and (5) phase 5, modulation of proinflammatory host immune response [245,248]. In immune compromised individuals: (1) the immunomodulatory activities of MSCs have raised safety concerns regarding the increased risk of viral infections and viral reactivation which are major causes of mortality following HSCT, and (2) the high susceptibility of MSCs to viral infections in vitro could reflect the destructive outcomes that might impair the clinical efficacy of MSC infusion [243]. However, data on the exact response of MSCs to viral infection in clinical settings is limited [243]. Examples of the immune regulatory properties of MSCs include: (1) inhibition of differentiation of monocytes to DCs, (2) alteration of cytokine profile of DCs resulting in down regulation of inflammatory cytokines and upregulation of regulatory cytokines, (3) induction of tolerant phenotypes of naïve and effector T-cells, (4) inhibition of antibody production by B-cells, and (5) suppression of natural killer (NK) cell proliferation and NK-mediated cytotoxicity [243]. BM-MSCs have emerging role in host defense: (1) they produce cytokines, chemokines and extracellular matrix (ECM) proteins, and (2) they may augment antimicrobial responses, abridge proinflammatory and damage responses, and ameliorate injury caused by the host defense to the pathogen [245,248]. The produced cytokines, chemokines and ECM proteins: support HSC survival and engraftment; influence immune effector cell development, maturation, and function; and inhibit alloreactive T-cell response [248]. So, BM-MSCs appear to function as a critical fulcrum providing balance by: promoting pathogen clearance during the initial inflammatory response, and suppressing inflammation to preserve host integrity and facilitate tissue repair [248].
MSCs could potentially be involved at multiple levels in host defense, assuming roles in hematopoiesis and mobilizing immune effector cells in: direct stimulation of pathogens, and modulation of proinflammatory immune responses so as to minimize the tissue damage induced by inflammation [248,257]. The immunomodulatory properties of MSCs are mediated by both: cell to cell interaction and the secreted cytokines [246,257]. Several studies have shown that the following cytokines are secreted by MSCs: IFN-γ; IFN-α; IL-6; IL-10; prostaglandin-E2; indoleamine-2,3-dioxygenase; TGF-β; vascular endothelial growth factor; intercellular adhesion molecule; CC chemokine ligand-2/monocyte chemotactic protein-1 (CCL-2/MCP-1); CCL-5/RANTES (regulated on activation, normal T-cell expressed and secreted); monocyte-CSF (M-CSF); GM-CSF; hepatocyte-growth factor; and other chemokines [243,246,247,249,258,262]. BM-MSCs may protect against infectious challenge either by direct effects on the pathogens or through indirect effects on the host [248]. BM-MSCs may reduce the burden of the pathogen either by inhibiting growth of the pathogen through soluble factors or by enhancing immune antimicrobial functions [248]. In the host, BM-MSCs may: attenuate proinflammatory cytokine and chemokine induction, reduce proiflammatory cell migration into sites of injury or infection, and induce immunomodulatory soluble and cellular factors to preserve organ function [248]. Additionally, MSCs can stimulate B-cell antibody production as they express miRNA for IL-6 which is important for B-cell differentiation and immunoglobulin secretion [263]. MSCs, particularly placenta-derived MSCs and fetal membrane-derived MSCs, are highly susceptible to herpes viruses including VZV [245,264].
Mesenchymal stem cells and blood brain barrier: Emerging strategies that can deliver therapeutic agents or cytotoxic genes to the brain across the BBB include: (1) nanoparticle carriers, (2) cell-based drug delivery via MSCs or neural stem cells (NSCs), and (3) focused ultrasound-based drug and gene delivery [265]. Studies have shown that several types of stem cells including BM-MSCs and NSCs can cross the BBB and reach tumors localized in the brain such as glioblastoma multiforme as well as ischemic areas and injured sites in the brain and engraft there. Hence, MSCs can be used as means of cellular carriers or Trojan horses to deliver cytotoxic genes or therapeutic agents for brain tumors, and they can be used to exert their therapeutic and regenerative effects in the brain [266-270]. In cancer, MSCs are a double-edged sword. They can exert stimulatory effects on tumor development and they can have inhibitory effects on cancer cell growth and metastases [271]. MSCs have anticancer properties as they can be engineered or modified to become carriers of suicide genes that can produce toxic products and subsequently target tumor cells and inhibit tumor expansion while keeping the surrounding healthy tissues intact [272,273]. Additional potential roles of MSCs in cancer therapeutics include: (1) MSCs can be employed as carriers of anti-angiogenesis factors that inhibit tumor growth and prevent metastases, (2) induction of cytokine gene expression in MSCs thus making tumor cells more exposed to the response of the host immune system, (3) antimitotic factors may become a rational target for MSC-based cancer engineering, (4) the use of exosomes as biological delivery vehicles for mRNA transfer as exosomes do not elicit acute immune rejection and do not have risk of tumor formation, and (5) targeting CSCs by engineered MSCs that can express TNF-related apoptosis inducing ligand [272,274].
Dendritic cells: DCs are BM-derived cells that arise from lympho-myeloid hematopoiesis and form an essential interface between the innate sensing of pathogens and the activation of adaptive immunity [275,276]. DCs are potent antigen presenting cells that are critical in the initiation of successful primary antiviral immune responses through stimulation of immunologically naïve T-cells to control and/or eliminate viral infections [276-280]. DCs are located in most tissues including the skin, blood, lymph and mucosal surfaces [276]. There are several classes or subsets of DCs. The 3 major subsets are: (1) plasmacytoid DC (pDC), (2) myeloid/conventional DC1 (cDC1), and (3) myeloid/conventional DC2 (cDC2), while other subsets include: (1) Langerhans cells, (2) pre-DC, (3) monocyte derived DCs, and (4) non-classical monocytes [275,280]. The specific subtypes of DCs are differentiated by an extended range of surface markers although it is difficult to dissociate cDC2 from monocyte-derived DC under certain circumstances [275,280]. Each subset of DCs develops under the control of a specific repertoire of transcription factors [275]. DC hematopoiesis is conserved between mammalian species and is distinct from monocyte development [275]. Functions of DCs include: inhibition and control of immune responses as well as bridging the innate and adaptive immune systems [275,280]. Immature DCs express the following: (1) MHC class I and MHC class II molecules, (2) CD 40 ligand, and (3) costimulatory molecules such as CD80 and CD86 [279]. Immature DCs have excellent antigen processing but poor antigen presenting capacity. When immature DCs are stimulated by CD 40 ligand, TNF-α, IFN-γ, and IL-6 they undergo a morphologic change and become mature DCs. Mature DCs express higher levels of certain markers such as CD 80 and CD 86. They also express CD 83 which increases their ability to stimulate T-cells. Mature DCs are excellent at both antigen processing and antigen presentation [279].
DCs are the Achilles heel of the immune system as they are essential for inducing antiviral immune responses [280-282]. DCs use the following 3 pathways to present antigens to CD8 and CD4 T-cells: (1) infection of DCs with viruses leads to expression of viral proteins that are processed into 8-10 amino acid peptides in the proteasome, (2) DCs can take up apoptotic virus-infected cells or viral proteins that are processed in endosomes, and (3) DCs can endocytose viral proteins or inactivated virus in vacuoles, process the proteins, load them on MHC class II molecules and then transport them to the cell surface for presentation to CD4 T-cells [279]. VZV infects both DCs and T-cells and exploits both as Trojan horses [281]. VZV exploits DCs to disseminate in the human body, evade the antiviral immune responses and cause disease [276]. DCs are required for generation of VZV-specific T-cells [281]. During primary infection: (1) VZV-infected DCs traffic to the draining lymph nodes and tonsils where the virus is transferred to the T-cells, and (2) VZV infected T-cells subsequently spread infection throughout the body to give rise to the typical skin eruption [276,281]. VZV can productively infect immature DCs, impair their function, and inhibit their maturation [278]. VZV inhibits NF-κβ signaling pathway in human DCs following protein phosphorylation but before translocation of NF-κβ subunits into the nucleus. The E3 ubiquitin ligase domain of ORF 61 is required for modulation of NF-κβ pathway [278]. VZV-ORF 47 is critical for replication of VZV in human immature, but not mature, DCs [279]. Mature DCs are permissive for VZV infection and DC infection can lead to transmission of the virus to T-lymphocytes in preparation for subsequent dissemination [276,277]. VZV infection of mature DCs reduces their ability to function properly thus VZV has evolved an immune evasion strategy that would likely impair immune surveillance and enhance the chance of life-long persistence in the human population [277]. VZV infection of mature DCs results in: (1) down-regulation of functionally important immune molecules such as: MHC class I, CD 80, CD 86, and CD 83; and (2) significant reduction in T-cell stimulatory capacity [277]. The induction of VZV-specific T-cell immunity is critical for host recovery from varicella [277]. Both MHC and class I-restricted CD8+ T-lymphocytes as well as class II-restricted CD4+ T-lymphocytes are sensitized to viral antigens during primary infection [276,277]. VZV has the capacity to interfere with the expression of MHC class I and MHC class II and this immunomodulatory mechanism plays an important role in the pathogenesis of VZV disease and persistence of the virus in the human host [276,277]. They also express CD 83 which increases their ability to stimulate T-cells [279,280]. Mature DCs are excellent at both antigen processing and antigen presentation [279].
Natural killer cells
NK cells develop from common progenitors and differentiate from HSCs in the BM but diverge into distinct subsets which differ in: cytokine production, cytotoxicity, homing and memory traits [283,284]. Sources of NK cells include: BM, peripheral blood, cryopreserved umbilical cord blood, human ESCs, iPSCs, and various cell lines such as NK-92 and KHYG-1 [285]. NK cells are large granular lymphocytes that are: CD3-, CD56+, CD16+, CD94+ and NKp46+ [285-287]. They can be classified into: (1) naïve CD56 bright CD 16 dim cells, and (2) mature CD56 dim CD16 bright cells [285,286,288]. Human NK cells have the following distinguishing features: expression of specific surface markers, intracellular signaling molecules and expression of transcription factors, tissue specific imprinting, and foreign antigen exposure [285,286,288,289]. NK cells are the third population of lymphoid cells but they represent the first line of defense against infections and tumors [285,288-292]. They have been traditionally classified as short-lived innate lymphocytes or part of the innate immune system because unlike T and B cells, NK cells do not express receptors that require gene rearrangements to generate receptor diversity and specificity [290]. Recently, it has been shown that NK cells exhibit many of the features associated with adaptive immunity such as: (1) the expansion of pathogen specific cells, (2) the generation of long-lasting memory cells that persist after cognate antigen encounter, (3) the ability to mount an enhanced secondary recall response to rechallenge, and (4) having distinct gene regulatory functions by adaptive NK cells [290,293]. Cancer cells frequently produce platelet derived growth factor receptor (PDGFR)-β which through autocrine and paracrine PDGFR-β signaling promotes: tumor growth, cell proliferation, metastasis, stromal recruitment, angiogenesis, and epithelial-mesenchymal transition [294].
Natural killer cells in cancer: NK cells play a major role in the immune response to certain malignancies by several mechanisms that include: (1) directly by secretion of potent immune mediators such as targeted secretion of cytokines or cytotoxic granules to cause cytolysis of transformed cells, (2) indirectly by orchestrating anti-tumor immune responses to prevent metastatic spread by engagement of the activating receptor NK p46 on NK cells, (3) the human immunoreceptor NK p44 expressed on NK cells and the innate lymphoid cells recognize PDGF-DD produced by tumor cells and this plays a major part in the control of tumor growth by NK cells, and (4) NK cells recruit conventional type I DCs into the tumor microenvironment to promote immune control of tumors [290,291,294-297]. Thus, NK cells play key roles in innate and adaptive responses through unique NK cell activation mechanisms during early host defense against viruses and tumors by performing 2 major roles: contact dependent cytotoxicity and cytokine production for immune modulation [284,290,295]. Target cell apoptosis is primarily mediated by perforin and genzyme B. The regulation of the immune responses is mediated by secretion of cytokines such as: IFN-γ, TNF-α, IL-1, IL-3, and GM-CSF [284,287]. NK cells are attractive candidates for adoptive cellular therapy in: (1) cancer: HMs such as acute leukemia, and solid tumors, with either CAR-engineered NK cells or combining NK cells with CD-16 binding antibodies or immune engagers; and (2) allogeneic HSCT including haploidentical allografts to protect against disease relapse by enhancing graft versus leukemia (GVL) effect without causing GVHD [12,283,285,286,297-300].
Natural killer cells and viral infections: NK cells play a major role in the immune response to certain viral infections by: (1) direct cytolysis or killing of virus-infected cells in order to rapidly control viral infection, and (2) secretion of potent immune mediators such as IFN-γ and other cytokines [287,290,301,302]. NK cells share features with long-lived adaptive immune cells and this can impact disease pathogenesis through inhibition of adaptive immune responses by virus-specific T and B cells as NK cells are potent regulators of antiviral T and B cell responses [301]. NK cells can produce persistent memory in response to certain viral infections particularly those caused by CMV [293]. NK cells have multiple mechanisms to kill virus infected cells through the engagement of extracellular death receptors, and through exocytosis of cytotoxic granules [292]. Mediation of cytolysis occurs through: engagement of death receptors expressed on target cells, and expression by NK cells of multiple extracellular ligands including fas ligand and TNF related apoptosis-induced ligand ultimately resulting in apoptosis of the target cells [292]. VZV actively manipulates the NK cell phenotype through productive infection. NK cells have a potential role in VZV pathogenesis and they are implicated in controlling infections caused by VZV [303]. Decreased NK cell function is associated with: (1) several genetic or hereditary disorders, (2) several chronic disorders such as: chronic fatigue syndrome, depression, autoimmune diseases, metastatic cancer, and exposure to occupational chemicals, and (3) certain viral infections such as HIV [287]. Although NK cell deficiencies are rare, they predispose to infections by herpes viruses [292]. VZV infects NK cells and causes: (1) cell to cell interaction with VZV-infected epithelial cells during early encounter or entry, and (2) subsequent modulation of NK cell function and phenotype resulting in stimulation of chemokine receptors and CD57 expression and inhibition of the expression of CD56, CD 16 and FcVRIII [292].
T-lymphocytes: T-cell mediated immunity consists of CD4 and CD 8 effector and memory T cells [304]. VZV-specific T-cells and T-cell mediated immunity are essential for controlling VZV infections [304,305]. Also, administration of varicella vaccine generates VZV-specific humoral and cellular immune responses [304]. However, VZV-specific T-cell mediated immunity decreases with immunosuppression and advancing age [306,307]. Studies in older subjects have shown: (1) differentiation of memory T-cells is defective after VZV vaccination, (2) the numbers of circulating IFN-γ secreting VZV-specific CD4+ T-cells are decreased, and (3) the number of foxp3+ regulatory T-cells and expression of the inhibitory receptor programmed cell death-1 on CD4+ T-cells are significantly increased in the skin [308,309]. Natural VZV infection and live-attenuated varicella vaccine elicit T-lymphocytes that recognize VZV glycoproteins (I-IV) and IE-62 protein [310]. Glycoproteins B and E are major targets of VZV-specific CD4+ and CD8+ T-cell recognition occurring during VZV infection in recipients of HSCT. Thus, glycoproteins B and E may form a basis for a novel non-hazardous VZV subunit vaccine that is suitable for immunocompromised HSCT patients [311]. VZV-specific T-cell immunity, which is essential to prevent VZV reactivation, can recover efficiently in recipients of T-cell depleted stem cell allografts to levels equivalent to those encountered in healthy virus carriers [312]. CD4+ cytotoxic T-cells are important in primary host response to acute varicella [313]. Immunization with live attenuated varicella vaccine can induce VZV-specific memory cytotoxic T-cell responses comparable to those occurring in individuals with normal or natural immunity [313].
HZ-DNA vaccines with IL-7 and IL-33 molecular adjuvants strongly elicit protective T-cell immunity [306]. The magnitude of VZV-specific CD4+ T-cell response increases after VZV vaccination [314]. CD4+ T-cell responses to SVV are more important than antibody or CD8+ T-cell responses in controlling primary SVV infection. Thus, eliciting robust CD4+ T-cell responses may enhance the efficacy of VZV vaccines [315]. Varicella vaccination is associated with increases in IE-62 peptide-specific CD8+ T-cell responses [316]. However, IE-62 protein is required for the initiation of VZV replication [305]. Antiviral cytotoxic T-lymphocyte activity against targets expressing VZV proteins is mediated equally by T-lymphocytes of CD4+ and CD8+ phenotype [317]. Infection with VZV induces cellular immunity that protects against reinfection and reactivation of the virus from neuronal sites of latency [318]. Memory T-cell recognition of VZV proteins has been characterized using in vitro methods to assess activation of CD4+ and CD8+ T-cells and induction of cytokine and cytotoxic T-lymphocyte responses [318]. In recipients of HSCT: (1) recognition of protective VZV-specific T-cell mediated immunity against VZV does not require disease development, and (2) monitoring of VZV-specific cell-mediated immunity can guide the initiation and cessation of antiviral prophylaxis [319].
Mononuclear cells: Infection of the immune cells by VZV results in attenuation of the antiviral mechanisms to control the infection and limit spread of the virus [320]. Several studies have shown that VZV productively infects human peripheral blood mononuclear cells (PBMNCs) and the infected PBMNCs then disseminate the virus to distal organs to produce clinical disease [320-324]. VZV induces an IFNmediated Th1 reaction in PBMNCs. Also, pDCs play an important role in IFN-α production during VZV infection through TLR9-dependent and-independent pathways [325]. Monocyte derived macrophages are also highly permissive to VZV infection [320]. However, growth of VZV in human adult monocytes is incomplete and restriction of VZV growth by monocytes may play a role in defense against VZV infection [326]. VZV-DNA can be detected by RT-PCR in human PBMNCs during viremia and within 1-23 days after onset of the skin lesions [323,324]. Several studies have shown that VZV-DNA can be detected by PCR in PBMNCs briefly during relapse in patients with multiple sclerosis and in patients with VZV infections experiencing PHN [327,329]. Also, VZV-DNA can be detected by in situ hybridization in PBMNCs in patients with VZV infection for 2-56 days after appearance of the skin eruption [330]. It is well recognized that TLRs are key components of the host innate recognition system and that TLR2 plays a role in the inflammatory cytokine production by monocytes during VZV infection [324]. VZV specifically induces IL-6 in human monocytes via TLR2-dependent activation of the NK-κβ signaling pathway. Unlike other herpes viruses, the cytokine response to VZV is species specific [324].
VZV proteins, cell components and cellular processes
Open reading frames: Approximately 80 proteins have been described to be produced in association with VZV infections: (1) 74 ORFs, 3 of them (ORF 62/71; ORF 63/70; ORF 64/69) are duplicated; (2) 3 glycoproteins: B, C and E; and (3) 3 IE proteins: IE 4, IE 62, and IE 63. Forty four of these ORF genes are essential for viral replication [46,48,101,311,331-333]. Additionally, VZV contains 5 unique ORF genes (ORF 1, ORF 2, ORF 13, ORF 32, and ORF 57) that are not present in HSV-1 and it lacks 15 ORF genes that are expressed by HSV-1 [48]. The most common ORFs are: ORF 1, ORF 2, ORF 4, ORF 10, ORF 13, ORF 21, ORF 23, ORF 29, ORF 32, ORF 47p, ORF 54, ORF 57, ORF 61, ORF 62, and ORF 63 [46,48,101,331,332]. One complete cycle of VZV replication that leads to a new generation of infectious VZV particles takes 9-12 hours [101]. Kinetic analysis of VZV replication cycle in individual fibroblasts has demonstrated the spatiotemporal expression of: (1) 6 VZV proteins [ORF61, ORF 62, ORF 63, ORF 29, ORF 23, and glycoprotein E]; (2) newly synthesized viral DNA; and (3) virion morphogenesis. However, IE 63 expression occurs at 4-6 hours which is later than that of IE 62 and ORF 61 [101]. Transcripts encoding 6 VZV genes (ORF 66, ORF 4, ORF 21, ORF 29, ORF 62, and ORF 63) have been detected in latently infected human as well as rodent ganglia as reported by several laboratories [334-336]. However, expression of the 2 latency-related VZV genes, ORF 62 and ORF 63, is regulated epigenetically through chromatin structure [335].
ORF 63 is expressed in an IE protein. It is present in the virion and has critical role in establishment of latency as it is one of the most abundant viral RNAs expressed during latency [335,336]. ORF 63 gene product is a tegument phosphoprotein with some regulatory functions such as enhancement of IE 62 transactivation of some VZV promoters [101]. Also, it is a prominent gene product in both productive and latent infection and may play a critical role in VZV pathogenesis by aiding neuron and keratinocyte survival [337]. ORF 61 and ORF 62 are expressed very early and this expression occurs less than one hour after VZV infection of human fibroblasts [101]. VZV encodes an IE protein termed ORF 61p which is a transcriptional activator of viral promoters [338]. ORF 61p enhances infectivity of viral DNA. Intact ORF 61p RING finger domain is necessary for E3 ubiquitin ligase activity and is required for autoubiquitination and regulation of protein stability [338]. ORF 21 is the first gene product expressed during latency [334]. Genome-wide mutagenesis has revealed that ORF 7 is a novel VZV skin-tropic factor which is essential for viral replication [339]. Application of enrichment protocols to targeted genome sequencing has revealed the unexpected deletion of a significant proportion of VZV-ORF 12 following propagation in culture human fibroblast cells [340]. ORF 25 gene product is an essential hub for protein interactions and is essential for VZV replication [341]. ORF 54 deletion mutant represents the first VZV encapsidation mutant that can serve as a platform for the isolation of portal mutants via recombination-mediated genetic engineering and can provide a strategy for more studies on VZV portal structure and function [331]. VZV-ORF 47 is critical for replication of the virus in immature, but not in mature, DCs and for spread of virus to other cells [279]. The protein coded by ORF 9, ORF 9p, is an essential tegument protein [332,342]. ORF 9p phosphorylation by ORF 47p is critical for the formation and egress of VZV viral particles. ORF 9p is essential for viral replication by binding to cellular adaptor protein complex 1 [342].
Glycoproteins: The lipid envelope of VZV contains numerous glycoproteins that are required for replication and pathogenesis [343]. VZV glycoprotein C activity facilitates the recruitment and subsequent infection of leukocytes, increases chemokine mediated leukocyte migration and hence enhances VZV systemic dissemination in humans [333]. Glycoproteins B and E are major targets of VZV-specific CD4+ and CD8+ T-cell reconstitution occurring during VZV infection or reactivation following allogeneic HSCT [311]. Glycoproteins B and E might form the basis for novel non-hazardous subunit vaccines suitable for immunocompromised hosts [311]. The cytoplasmic domain lysine cluster of VZV glycoprotein B is implicated in the regulation of glycoprotein B fusion, ultimately leading to attenuation of VZV infection if unmodulated, so this domain is critical for the regulation of VZV cell to cell fusion and VZV infection [343,344]. VZV glycoprotein M is important for efficient cell to cell virus spread but it is not essential for virus growth [345]. A site of vulnerability which has been identified by structural studies of neutralizing antibodies is bound to the glycoprotein complex gHgL [346].
Promyelocytic leukemia protein: Recently, the following cellular proteins: promyelocytic leukemia protein (PML), hDaxx, and Sp100 have been identified [100,347]. These cellular proteins form a subnuclear structure called nuclear domain 10 (ND10) or PML nuclear bodies as host restriction factors that counteract herpes viral infections by inhibiting viral replication at different stages [347]. The antiviral function of ND10 is antagonized by viral regulatory proteins, such as IE1, which induce either a modification or disruption of ND10 [347]. PML is an essential regulator of somatic cell programming and stem cell pluripotency [348]. PML has diverse functions that regulate: response to DNA damage, apoptosis, senescence, and angiogenesis [349,350]. PML is a regulator of metabolic pathways in stem cell compartments including hematopoietic system and it has provided new strategies for controlling stem cell maintenance and differentiation [349]. For its action, PML recruits other proteins such as: Sp100, Daxx, small ubiquitin-like modifier (SUMO)-1, CBP, and P53 [350]. PML nuclear bodies are matrix-associated domains that recruit a variety of unrelated proteins. PML-NBs are found in most cell lines and many tissues and are regulated by cellular stresses such as: DNA damage, viral infection, transformation, and oxidative stress [351]. The PML protein is a key organizer of large numbers of proteins that share the ability to be SUMOylated [351]. Human PML is located on chromosome 15 and has 9 exons and ≥ 11 isoforms [352]. PML is an IFN-inducible protein that is involved in restricting VZV replication [352]. PML-isoform IV acts at the following 2 stages that require PML IV SUMOylation: (1) it confers viral resistance directly in an IFN-independent manner, and (2) it specifically enhances IFN-β production ultimately protecting uninfected cells from ongoing viral infection [353]. IFNs are the first line of defense against viral infections [353]. More recently, there is a growing body of evidence supporting the impression that PML is a key regulator of cytokine signaling [354]. PML is involved in various cellular processes including: cell death, senescence and antiviral defense and it is able to interact with various partners either in the cytoplasm or in the nucleus [354].
Chaperons: Chaperons are a diverse group of molecular proteins that function during homeostasis and stress conditions such as disease or infection in all living organisms [355]. Chaperons play critical roles in: (1) assisting folding and refolding of protein chains, (2) assisting protein transport and translocation through membranes, (3) facilitating degradation of proteins, (4) involvement in host-pathogen interaction during infection leading to the development of innate and adaptive immunity, and (5) involvement in protein quality control [355,356]. Currently, ELISA-based tests are used to measure the plasma levels of chaperons. However, these tests give information about the quantity or amount but not the function or activity of chaperons [355]. Chaperons are large cylindrical complexes that provide a central compartment for a single protein chain to fold unimpaired by aggregation [357]. Protein folding, maintenance of proteome integrity and protein homeostasis or proteostasis clinically depend on a complex network of molecular chaperons. Additionally, folding of proteins occurs upon controlled release of newly synthesized proteins from these factors or after transfer of downstream chaperons such as the chaperonins [357].
Molecular chaperons are required for the folding processes of many proteins [358]. The core chaperone machinery consists of chaperonins and heat shock proteins. The chaperon machinery is maintained from prokaryotes to eukaryotes although the chaperon network is expanded in eukaryotes [358]. Proteins with RNA chaperon activity have diverse structures and functions and they play important roles in cellular mechanisms [359]. Chaperon rings play a vital role in the opposing ATP-mediated processes of folding and degradation of many cellular proteins through incompletely understood mechanisms [360]. The cell protein BAG3, a host chaperon, is specifically required for efficient replication of VZV and not HSV [361]. Alteration of host chaperon activity provides a novel means of regulating viral replication. VZV-ORF29p, a single stranded DNA binding protein, is one of the VZV-latency associated proteins [361]. Activating transcription factor 3 is a host factor that plays a role in maintaining latent HSV infection in the sensory neurons of murine trigeminal ganglia [362]. Virus-induced chaperone-enriched domains are nuclear protein quality control centers that are produced during early HSV-1 infection and are mediated by the virus to promote productive infection [356]. VZV glycoprotein (gL) is a simpler form of gL chaperone protein than EBV-gL glycoprotein [363]. Targeting viral enzymes, proteins and chaperones may become a new therapeutic modality for treating infections caused by drug resistant herpes viruses [364,365].
SUMO proteins and SUMOylation: Viruses alter specific host cell targets, through various mechanisms, to counteract host defenses aimed at eliminating infectivity and viral propagation [366-368]. Post-translational modification (PTM) of proteins is important to numerous cellular events thus allowing cells to respond to both internal as well as external stimuli [367]. The most studied protein modifications include: ubiquitination, phosphorylation, acetylation, methylation, and glycosylation [367]. PTMs contribute to signaling pathways such as: gene regulation, epigenetics, differentiation, protein degradation, and tumorigenesis [369]. SUMOylation was first described in the year 1996, then in 1997 a new type of modifying protein (SUMO) was identified and since that time 4 SUMO isoforms; SUMO-1, SUMO-2, SUMO-3, and SUMO-4; have been characterized in humans [366368]. SUMO proteins are essential for the normal function of all eukaryotic cells [370,371]. The SUMOylation process begins with the transcription and translation of the SUMO genes to produce the SUMO pro-peptide [367]. SUMOylation is a highly conserved and reversible PTM that is manipulated by viruses in order to modulate: anti-viral responses, viral replication and viral pathogenesis [366-370]. SUMOylation is a major regulator of protein function that plays an important role in a wide range of cellular processes [369,370]. SUMOylation is carried out by a cascade of several enzymatic steps where SUMOs are implicated in the regulation of diverse cellular processes particularly nuclear events [367,372]. Thus, SUMOylation is an important mechanism regulating the activities of various proteins involved in cellular processes such as: DNA replication and repair, chromosome packing and dynamics, genome integrity, nuclear transport, signal transduction, and cell proliferation [373]. SUMO-specific proteases are required for the maturation of SUMO precursors and to reverse a wide range of cellular processes [367,369]. There are 4 SUMO paralogues and an increasing number of proteins are being identified as SUMO substrates [367,370]. SUMO Ubc9 enzyme represents a leading target for viral proteins and an attractive biomarker in the treatment of most viral-induced human pathologies [366]. SUMOylation controls the function of several proteins and biological processes [368]. SUMOylation, which has a central regulatory role in PTM, is widely exploited by viruses [368]. A number of viral proteins either modify or become modified by the SUMOylation system in order to create a cellular environment that favors their survival and propagation and prevents host antiviral responses [368].
ORF 61p of VZV is important in the infectivity of the viral DNA. ORF 61p appears to target substrates for potential degradation in a SUMO-independent manner. It has much stronger affinity for SUMO-1 than SUMO-2 and SUMO-3 [368,369,371]. Additionally, it uses substrate binding sites other than SIMs to recognize some of its substrates. Suppression of nuclear factor kappa β (NF-κβ) activation by TNF-α requires a functional RING domain of ORF 61p but not it’s SIMs [368,369,371]. VZV-ORF 29 gene encodes a single-stranded DNA binding protein that is predominantly nuclear during lytic infection but appears to be restricted to the cytoplasm of latently infected neurons [374]. ORF 29p can be ubiquitinated and SUMOylated but these processes implicate the proteasome as one of the determinants of the protein’s cell type-specific localization [374]. Tripartite mofit (TRIM) proteins have been implicated in multiple cellular functions including antiviral activity. The antiviral activity of TRIMs relies mainly on their function as E3-ubiquitin ligases [371,375]. Several TRIM family members mediate innate immune cell signal transduction and subsequent cytokine induction. E3-ubiquitin ligases can recognize SUMOylated proteins and link SUMO modification to the ubiquitin/proteasome system [371,375]. Advanced investigations of ubiquitiylation and SUMOylation during virus-host interaction have shown that human viruses have evolved a large arsenal of strategies to exploit specific PTM processes [369]. Identification and knowledge of virus-mediated PTM manipulation by viral analogs infiltrating ubiquitin/SUMO pathways will provide valuable information for the development of future antiviral drugs and novel immunotherapies [366,367,369]. So, targeting SUMOs could represent a new therapeutic strategy against viral infections [376].
MicroRNAs: Micro-RNAs (miRNAs) were first discovered by Ambros and colleagues in 1993 [377]. miRNAs have several roles or functions that include: (1) regulation or modulation of gene expression; (2) downregulation of target protein expression in cells; (3) regulation and maintenance of numerous cellular physiological functions or processes such as immune function, apoptosis, and tumor genesis; (4) regulation of interaction between hosts and viruses; and (5) inhibition of viral replication [377-380]. VZV encodes several miRNAs or small non-coding RNAs that regulate VZV infection in host cells [379]. Studies, using quantitative RT-PCR, have shown several circulating miRNAs in patients with VZV infection including: miR-197; miR-629; miR-363; miR-132; miR-122 as well as miR-1906; miR-571; miR1276; miR-1303; miR-943; and miR-661 [378,381]. Hence, circulating miRNAs can be potentially used as biomarkers of active or latent VZV infection [378,381]. RNA sequencing analysis has shown that VZV infection has profound effect on differentiating keratinocytes thus promoting blistering and desquamation of the skin through upregulation of kallikreins and serine proteases and alteration of epidermal gene expression [382]. Regulation of gene transcription in VZV requires coordinated binding of multiple host and virus proteins onto specific regions of the virus genome [383]. Hyperphosphorylation of serine 5 of the c terminal domain of phosphorylated RNA polymerase II (RNAP) along with the lengths of VZV genes is independent of miRNA abundance as RNAP is associated with human and virus transcriptional units through different mechanisms [383].
Extracellular vesicles and exosomes
Extracellular vesicles: Extracellular vesicles (ECVs) are nano-sized, cell-derived particles or membrane surrounded structures that are released by most cell types [256,384-386]. ECVs are potent vehicles of intercellular communication to transmit biological signals between cells and they are characterized by a specific set of proteins, lipids and nucleic acids [384,386]. The 4 main types of ECVs are: exosomes which are the most widely studied type of ECVs, microvesicles, apoptotic bodies, and oncosomes [256,385-387]. Classification of ECVs is based on their bio biophysiological properties including: size, cellular origin, molecular cargo, and biogenesis. ECVs can be isolated from various biological fluids including: blood, urine, cerebrospinal fluid, amniotic fluid, seminal fluid, and breast milk. Interestingly, they were initially considered as mostly cellular debris [385,386]. ECVs are the key mediators of intercellular communication by: reprogramming target cells, and regulating numerous physiological processes and pathological conditions such as: cancer, inflammation and immune response, and viral infections [256,385,386]. During viral infection, ECVs have 2 key biological activities: (1) transport of viral genomes into target cells, and (2) intervention in cell physiology to facilitate viral infection [385]. MSC-derived ECVs may provide a new therapeutic option in cell transplantation or gene therapy for different diseases particularly HMs [256,384]. Also, they may have therapeutic effects in immune regulation, tumor inhibition, and regenerative medicine [256].
Exosomes: Exosomes are ECVs that originate as intraluminal vesicles during the process of multivesicular body formation [388]. The contents of exosomes are variable and depend on the cells from which they originate and they include: proteins, miRNAs, lipids, and carbohydrates [389]. Exosomes were first described in the early 1980s and in the late 1980s the word “exosomes” was coined by Rose Johnstone [388]. Exosomes mediate intercellular communication through functional or biologically active proteins, lipids, and RNAs [388]. Several studies have demonstrated that exosomes are implicated in: (1) normal physiological processes such as modulation of the immune system, metabolism, and neural development and (2) progression of several pathologies such as: cancer, infection and neurodegeneration [386].
Exosomes released from virus-infected cells contain various viral and host cellular factors that can modify the host cell responses [388]. They allow the host to produce effective immunity against pathogens by activating antiviral mechanisms and by transporting antiviral factors between cells. Therefore, exosomes are crucial components in the pathogenesis of viral infections [388]. Viruses including herpes viruses can manipulate exosomal pathways [389]. VZV could utilize the alterations in host exosomes to: enhance spread of the virus, evade host immune surveillance, and elicit pathological effects within the host [390]. Exosomes are emerging as critical mediators of viral infection-associated intercellular communication and microenvironment alterations [387]. Exosomes represent a source of viral infection that can be used as: (1) biomarker of disease and (2) target for therapy in order to control or even eradicate viral infection [387,389]. They can help to enhance immune responses of the host against pathogens by activating antiviral mechanisms. Thus, exosomes can be used as therapeutic agents to modulate immune responses [388]. Exosomes are prone to viral exploitation. For example, herpes viruses can hijack host exosomes for viral pathogenesis, that is, herpes viruses can hijack exosomal pathways to ensure their survival and persistence [97,387,388,391]. Herpes viruses utilize ECVs including exosomes for the complex interplay between infected host and recipient cells. These viruses incorporate genome expression products and direct cellular products into exosomal cargoes [387].
Role of cytokines in VZV infections
Cytokines are low molecular weight extracellular polypeptides or glycoproteins that are synthetized by different immune cells such as T-cells, neutrophils and macrophages in response to infection, inflammation or trauma [392]. There are different types of cytokines and these types include: chemokines, lymphokines, IFNs, ILs, and TNF [392]. Cytokines are important mediators of immune response and they are responsible for promotion and regulation of various immune responses [392,393]. Actions of cytokines can be categorized into: (1) autocrine: exhibited in the sites where cytokines are produced, (2) paracrine: shown in nearby cells, and (3) endocrine: exhibited in distant cells [392]. Cytokines play an essential role in the expression of cell mediated immunity as they facilitate cellular behavior in the development of an immune response [393]. During infection, herpes viruses produce various cytokines and chemokine receptors from genes in the viral genome [394]. They also regulate several host cellular genes to direct cellular chemotaxis for the benefit of the invading viruses. Consequently, viral-induced chemotaxis contributes to the epidemiology and persistence of herpes viruses [394]. The high prevalence of herpes viruses in humans is principally due to their ability to establish life-long latency and their exceptional capacity to modulate the host immune responses [395]. Functions of the chemokine components of herpes viruses include: attraction of target cells, blockade of leukocyte migration, and modification of gene expression and cell entry by the viruses [395].
Studies have shown that: (1) the transcription factor NF-κβ is a key factor in inducing the expression of IFN and other proinflammatory cytokines that are involved in antiviral response, and (2) effector proteins are involved in: inhibition of protein synthesis, degradation of RNA, modulation of viral genome, activation of the innate immune system, and development of the adaptive immune response [396]. Human herpes viruses deploy viral chemokines and viral chemokine receptors as well as chemokines and chemokine receptors pirated from the host to exploit and evade the host immune response [395]. Cells respond to a variety of cytokines and chemokines that allow them to migrate in different body locations depending on where they are required [394]. VZV could utilize glycoproteins as chemo-attractants to induce migration of PMNLs [394]. On the first infection with VZV, CD4 and CD8 T-cells are induced and this is followed up by generation of VZV IgM, IgG, and IgA antibodies, while memory immunity to VZV is characterized by persistence of: IgA antibodies, CD4 helper T-cells and cytotoxic T-cells within CD4/CD8 subpopulation [397,398]. Several studies have shown that the following cytokines are expressed or elevated in the serum following VZV infections: Il-6, IL-10, IL-8, IL-17, IL-4, IL-12, IL-21, IL23, and IL-1β in addition to IFN-α and IFN-γ [393,324,397-405]. Expression of IFN-α and IFN-β is upregulated in the early phases of VZV infection then IFN expression decreases significantly during the late phases of VZV infection [398,401]. In patients with PHN, which is a common complication of HZ infection, the serum levels of autoantibodies against: IFN-α, IFN-γ, GM-CSF, and IL-6 have been found to be markedly elevated [406]. IL-6 in particular may be associated with the development of PHN in the acute phase of HZ infection [407].
Additionally, IL-6 and IFN-γ may be used as early serum markers for the development of complications of VZV disease such as skin infection [408]. VZV infection of primary spinal cord astrocytes causing myelopathy and VZV infection of primary hippocampal astrocytes that causes encephalopathy have produced distinctive alterations and patterns of proinflammatory cytokine suppression that could contribute to the ineffective viral clearance in both VZV associated myelopathy and encephalopathy [409]. Also, patients with encephalopathy have shown elevated cerebrospinal fluid (CSF) levels of the following matrix metalloproteases (MMPs): MMP-3, MMP-8 and MMP-12 and the pronounced increase in CSF levels of MMPs correlated with severity of the disease while patients with meningitis have shown significant increase in CSF levels of MMP-9 [410]. In patients with VZV associated vasculopathy and giant cell arteritis (GCA), the following cytokine alterations have been reported: (1) upregulation of IL-6 which is the major cytokine involved in GCA, (2) upregulation of IL-6 and VEGF-A that can be utilized as marker of disease progression, while (3) programmed death-ligand 1 is downregulated in order to promote persistence of inflammation [400]. ARN and posterior uveitis represent severe intraocular infections that may be complicated by: retinal detachment, occlusive vasculitis, and loss of vision. VZV infection is the commonest cause of both of these eye inflammatory disorders [411,412]. Studies have shown that the following cytokines are significantly elevated during ARN: IL-6, IL-8, IL-10, IL-18, IL-15, MIF, MCP-1, Eotoxin, IP10, sICAM-1, and sVCAM-1 and their levels correlate with disease activity, while low levels of the following cytokines have been encountered: IL-2, IL-4, IL-13, and IFN-α [411,412]. IFN-γ release assay may be useful as a surrogate test for measuring VZV-specific immunity [413]. Also, characterization of cytokine, chemokine and growth factor responses during different stages of VZV infection may facilitate the development of effective immunotherapeutic as well as vaccine strategies [414].
Signaling pathways
Signaling pathways involved in VZV infections: Several signaling pathways are involved or activated in VZV infections and these include: (1) Janus kinase/signal transducer and activation of transcription (JAK/STAT) pathway; (2) c-Jun N-terminal Kinase (JNK) pathway; (3) extracellular signal-regulated kinase (ERK/MEK) pathway; (4) phosphatidylinositol 3-kinase (PI3K/Akt) pathway; (5) NK-κβ pathway; (6) mitogen-activated protein kinase (MAPK) pathway; (7) Wnt-Wingless pathway; and (8) cyclic-AMP response element binding protein (CREB) pathway. However, JAK/STAT signaling is the most studied pathway [278,415-422].
IFNs and JAK/STAT pathway: The induction of the JAK/STAT pathway by IFNs leads to the upregulation of hundreds of IFN-stimulated genes, many of which have the ability to rapidly kill viruses within infected cells [423]. During their evolution, viruses have acquired an extraordinary range of strategies to counteract the host immune responses by targeting the JAK/STAT signaling pathway in particular. Thus, almost all viruses have evolved strategies to combat the effects of type 1 and type 3 IFNs by antagonizing the IFN signaling pathways [423]. STAT 3 regulates several biological functions including: cell growth and proliferation, cellular differentiation, cell survival and apoptosis, angiogenesis, chemotaxis and cell adhesion, and immune response. STAT3 plays a key role in regulating immune and inflammatory responses of the host and the pathogenesis of many cancers [424-426]. IFNs are a multifunctional family of cytokines that play a critical role as first line defense against viral infections [423]. Type I IFNs (α/β) are induced in virus infected cells by engagement of viral molecules particularly nucleic acids with pattern recognition receptors to induce an innate immune response [423,427]. IFN-γ (type II IFN) is mainly produced by NK cells and T-cells. VZV downregulates STAT1 and JAK2 protein levels in virus-infected cells [428]. Studies have shown that: (1) SVV targets 3 proteins in the IFN-γ signal transduction pathway to escape the antiviral effects of IFN-γ, and (2) both SVV and VZV encode multiple gene products that tightly control IFN-induced antiviral responses [415,428].
However, ORF-63 prevents induction of IFN stimulated genes both directly and indirectly [415]. Several studies have reported differential regulation of STAT3 in a range of viral infections. STAT3 directs apparently contradictory responses to viral infections as it exhibits proviral as well as antiviral roles [424]. STAT3 activation and upregulation of survivin is a common and an important mechanism in the pathogenesis of lytic and tumorigenic herpes viruses. Also, STAT3 activation is critical for the life cycle of VZV because VZV skin infection is necessary for viral transmission and persistence in humans [426]. STAT3 is a key regulator in inflammation and tissue regeneration triggered by almost every pathogenic infection [425]. Viruses must deal with the STAT3 activity by either curtailing it or employing it [425]. Survivin, which is abundant in cancers and tissues that contain proliferating cells, mediates a necessary virus-enhancing effect of STAT3 activation on VZV [426]. The major IE-62 protein of VZV is delivered to the newly infected cell nuclei where it initiates VZV replication [429]. IE-62 protein is the predominant VZ virion tegument protein [427]. It activates the expression of all kinetic classes of VZV genes [427,429]. IFN-γ blocks VZV replication by inhibiting IE-62 function in a cell line-dependent manner [429]. IFN-α exerts multiple inhibitory activities against VZV infections as it inhibits VZV replication by several mechanisms [427]. Targeting IE-62 protein may offer a novel approach to the development of antiviral agents against VZV infections [427]. Identification of factors, encoded by viruses, which modulate the JAK/STAT pathway have opened new opportunities to develop new vaccines as well as antiviral agents [417].
Other signaling pathways in VZV infection
Herpes viruses can manipulate and control the Wnt/β-catenin signaling pathway to: promote viral replication, evade host immune recognition, and maintain latency. However, the involvement of this pathway during VZV infection has been under investigated [430]. VZV optimizes the conditions of its replication by: upregulation of proviral systems, and suppression of potentially antiviral-acting systems. For example, the ERK/MEK signaling pathway is influenced by VZV [416]. The PI3K/Akt signaling pathway has an essential role in successful replication of VZV [418]. JNK pathway plays an important role in lytic infection and reactivation of VZV in physiologically relevant cell types and may provide an attractive target of antiviral therapy. Also, MAPKs play a role in VZV infection of non-neural cells with distinct consequences for infectivity in different cell types [417]. ORF-61 has an important role in the regulation of MAPK signaling pathway and in VZV gene expression [422]. CREB is a factor involved in the regulation of several cellular processes. It is activated upon infection of T-cells with VZV. CREB activation is important for VZV skin infection and may be targeted by new antiviral drugs [419]. E3 ubiquitin ligase domain of ORF-61 is required to modulate NF-κβ signaling pathway. VZV has been found to inhibit the NF-κβ signaling pathway in human DCs [278]. Apparently, a large number of different elements including BM stromal cells, immune cells, cellular proteins, extracellular vesicles, cytokines, chemokines, ligands, as well as signaling pathways are involved not only in the pathogenesis, but also in the consequences of VZV infections. The various elements that are implicated in VZV infections are illustrated in figure 3. [101,204-207,245,248,268,272-274,276,292,304-307,310-313,320-324,334,341,343-346,349,350,361,368-371,378,381,387,397-405,415-422].
Figure 3: The various elements involved in the pathogenesis and consequences of VZV infections. VZV: Varicella Zoster Virus, ORFs: Open Reading Frames, PML: Promyelocytic Leukemia Protein, SUMOs: Small Ubiquitin-Like Modifier, MSCs: Mesenchymal Stem Cells, DCs: Dendritic Cells, NKCs: Natural Killer Cells, MNCs: Mononuclear Cells, miRNAs: micro-RNAs.
As clearly shown in the review, VZV differs from other herpes viruses and has the following peculiar features: having the smallest genome; losing almost all the genes that are not essential for its survival; being highly cell-associated; having no inhibitors of autophagy; being an exclusively human pathogen; having a species-specific cytokine profile; having inverse relationship with glioma; association with GVHD in recipients with HSCT; in addition to having BM stimulatory effects as well as several antitumor actions in patients with BM failure and HMs. The reported beneficial effects of VZV are rather outstanding and they have shown clear evidence of stimulation of BM activity and superior survival outcome in immune compromised patients infected with VZV. This should encourage scientists and researchers to give this potentially useful virus the attention it deserves. The positive effects of VZV on BM activity and on diseases such as BM failure syndromes, HMs and solid tumors through direct and indirect immunological mechanisms merit thorough investigations. The virus itself or constituents obtained from the serum of patients infected with VZV may ultimately become extremely valuable therapeutic modalities in the management of patients with various BM failure, HMs and solid tumors. Explanation of the stimulatory effect exerted on the 3 cell lines in the BM that is subsequently translated into increases in all blood counts as well as the antitumor effects of the virus can be explained by one or more of the mechanisms outlined or may be due to a new mechanism yet to be elucidated.
- Al-Anazi KA, Al-Jasser AM, Evans DA. Effect of varicella zoster virus infection on bone marrow function. Eur J Haematol. 2005; 75: 234-240. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16104880
- Kamber C, Zimmerli S, Suter-Riniker F, Mueller BU, Taleghani BM, et al. Varicella zoster virus reactivation after autologous SCT is a frequent event and associated with favorable outcome in myeloma patients. Bone Marrow Transplant. 2015; 50: 573-578. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25599166
- Al-Anazi KA, Kanfar S, Aldayel A, Abduljalil O, Sayyed AH. Reversal of pure red cell aplasia by varicella zoster virus infection. J Hematol Clin Res. 2019; 3: 1-10.
- Olkinuora H, von Willebrand E, Kantele JM, Vainio O, Talvensaari K, et al. The impact of early viral infections and graft-versus-host disease on immune reconstitution following paediatric stem cell transplantation. Scand J Immunol. 2011; 73: 586-593. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21323694
- Matsumura-Kimoto Y, Inamoto Y, Tajima K, Kawajiri A, Tanaka T, et al. Association of cumulative steroid dose with risk of infection after treatment for severe acute graft-versus-host disease. Biol Blood Marrow Transplant. 2016; 22: 1102-1107. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26968790
- Fuji S, Kapp M, Einsele H. Possible implication of bacterial infection in acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Front Oncol. 2014; 4: 89. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24795865
- Olkinuora HA, Taskinen MH, Saarinen-Pihkala UM, Vettenranta KK. Multiple viral infections post-hematopoietic stem cell transplantation are linked to the appearance of chronic GVHD among pediatric recipients of allogeneic grafts. Pediatr Transplant. 2010; 14: 242-248. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19691523
- Talmadge JE. Hematopoietic stem cell graft manipulation as a mechanism of immunotherapy. Int Immunopharmacol. 2003; 3: 1121-1143. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12860168
- Orti G, Barba P, Fox L, Salamero O, Bosch F, et al. Donor lymphocyte infusions in AML and MDS: enhancing the graft-versus-leukemia effect. Exp Hematol. 2017; 48: 1-11. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28027963
- Villa N Y, Rahman M M, McFadden G, Cogle C R. Therapeutics for graftversus-host disease: from conventional therapies to novel virotherapeutic strategies. Viruses. 2016; 8: 85.
- Xia G, Truitt RL, Johnson BD. Graft-versus-leukemia and graft-versus-host reactions after donor lymphocyte infusion are initiated by host-type antigenpresenting cells and regulated by regulatory T cells in early and long-term chimeras. Biol Blood Marrow Transplant. 2006; 12: 397-407. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16545723
- Cruz CR, Bollard CM. T-cell and natural killer cell therapies for hematologic malignancies after hematopoietic stem cell transplantation: enhancing the graftversus-leukemia effect. Haematologica. 2015; 100: 709-719. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26034113
- Dutt S, Baker J, Kohrt HE, Kambham N, Sanyal M, et al. CD8+CD44(hi) but not CD4+CD44(hi) memory T cells mediate potent graft antilymphoma activity without GVHD. Blood. 2011; 117: 3230-3239. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21239702
- Weber G, Gerdemann U, Caruana I, Savoldo B, Hensel NF, et al. Generation of multi-leukemia antigen-specific T cells to enhance the graft-versus-leukemia effect after allogeneic stem cell transplant. Leukemia. 2013; 27: 1538-1547. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23528871
- Chang YJ, Zhao XY, Huang XJ. Strategies for enhancing and preserving antileukemia effects without aggravating graft-versus-host disease. Front Immunol. 2018; 9: 3041. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30619371
- Dickinson AM, Norden J, Li S, Hromadnikova I, Schmid C, Schmetzer H, et al. Graft-versus-leukemia effect following hematopoietic stem cell transplantation for leukemia. Front Immunol. 2017; 8: 496. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28638379
- De Girolamo L, Lucarelli E, Alessandri G, Avanzini MA, Bernardo ME, et al. Italian Mesenchymal Stem Cell Group. Mesenchymal stem/stromal cells: a new ''cells as drugs'' paradigm. Efficacy and critical aspects in cell therapy. Curr Pharm Des. 2013; 19: 2459-2473. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23278600
- Auletta JJ, Eid SK, Wuttisarnwattana P, Silva I, Metheny L, et al. Human mesenchymal stromal cells attenuate graft-versus-host disease and maintain graft versus-leukemia activity following experimental allogeneic bone marrow transplantation. Stem Cells. 2015; 33: 601-614. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25336340
- Kawano N, Gondo H, Kamimura T, Aoki K, Iino T, et al. Chronic graft-versushost disease following varicella-zoster virus infection in allogeneic stem cell transplant recipients. Int J Hematol. 2003; 78: 370-373. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14686497
- Raymond AK, Sing letary HL, Nelson KC, Sidhu-Malik NK. Dermatomal sclerodermoid graft-vs-host disease following varicella-zoster virus infection. Arch Dermatol. 2011; 147: 1121-1122. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21931063
- Baselga E, Drolet BA, Segura AD, Leonardi CL, Esterly NB. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996; 23: 576-81. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9001991
- Weisdorf D, Zhang MJ, Arora M, Horowitz MM, Rizzo JD, et al. Graft-versushost disease induced graft-versus-leukemia effect: greater impact on relapse and disease-free survival after reduced intensity conditioning. Biol Blood MarrowTransplant. 2012; 18: 1727-1733. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22766220
- Yeshurun M, Weisdorf D, Rowe JM, Tallman MS, Zhang MJ, et al. The impact of the graft-versus-leukemia effect on survival in acute lymphoblastic leukemia. Blood Adv. 2019; 3(4): 670-680. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30808685
- Negrin RS. Graft-versus-host disease versus graft-versus-leukemia. Hematology Am Soc Hematol Educ Program. 2015; 2015: 225-230.
- Belcaid Z, Lamfers ML, van Beusechem VW, Hoeben RC. Changing faces in virology: the Dutch shift from oncogenic to oncolytic viruses. Hum Gene Ther. 2014; 25: 875-884. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25141764
- Rudd PA, Herrero LJ. Viruses: friends and foes. In: Cartilage repair and regeneration. Intech Open. 2017.
- Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016; 107:1373-1379. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27486853
- Ajina A, Prospects for combined use of oncolytic viruses and CAR Tcells. J. Immunother Maher J. Cancer. 2017; 5: 90. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29157300
- Howells A, Marelli G, Lemoine NR, Wang Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front Oncol. 2017; 7: 195. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28944214
- Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, et al. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013; 119: 421-475. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23870514
- Chaurasiya S, Chen NG, Warner SG. Oncolytic virotherapy versus cancer stem cells: a review of approaches and mechanisms. Cancers. 2018; 10: 124. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29671772
- Forbes NS, Coffin RS, Deng L, Evgin L, Fiering S, et al. White paper on microbial anti-cancer therapy and prevention. J Immunother Cancer. 2018; 6: 78. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30081947
- Wennier ST, Liu J, McFadden G. Bugs and drugs: oncolytic virotherapy in combination with chemotherapy. Curr Pharm Biotechnol. 2012; 13: 1817-1833. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21740354
- Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013; 12: 103. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24020520
- Ma W, He H, Wang H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018; 19: 40. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30563466
- Irwin CR, Hitt MM, Evans DH. Targeting nucleotide biosynthesis: a strategy for improving the oncolytic potential of DNA viruses. Front Oncol. 2017; 7: 229. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29018771
- Zhao C, Wang M, Cheng A, Yang Q, Wu Y, et al. Programmed cell death: the battlefield between the host and alpha-herpesviruses and a potential avenue for cancer treatment. Oncotarget. 2018; 9: 30704-30719. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30093980
- Amirian ES, Scheurer ME, Zhou R, Wrensch MR, Armstrong GN, et al. History of chickenpox in glioma risk: a report from the glioma international casecontrol study (GICC). Cancer Med. 2016; 5: 1352-1358.
- Canniff J, Donson AM, Foreman NK, Weinberg A. Cytotoxicity of glioblastoma cells mediated ex vivo by varicella-zoster virus-specific T cells. J Neurovirol. 2011; 17: 448-454. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21792750
- Pundole X, Amirian ES, Scheurer ME. Role of varicella zoster virus in glioma risk: Current knowledge and future directions. OA Epidemiology. 2014; 2: 6.
- Leske H, Haase R, Restle F, Schichor C, Albrecht V, et al. Varicella zoster virus infection of malignant glioma cell cultures: a new candidate for oncolytic virotherapy? Anticancer Res. 2012; 32: 1137-1144. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22493342
- Kennedy PGE, Gershon AA. Clinical features of varicella-zoster virus infection. Viruses. 2018; 10: 609. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30400213
- Weber DJ. Prevention and control of varicella-zoster virus in hospitals. Up To Date. 2019.
- Cohrs RJ, Gilden DH, Mahalingam R. Varicella zoster virus latency, neurological disease and experimental models: an update. Front Biosci. 2004; 9: 751-762. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14766405
- Tombácz D, Prazsák I, Moldován N, Szűcs A, Boldogkői Z. Lytic transcriptome dataset of varicella zoster virus generated by long-read sequencing. Front Genet. 2018; 9: 460. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30386374
- Sommer MH, Zagha E, Serrano OK, Ku CC, Zerboni L, et al. Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J Virol. 2001; 75: 8224-8239. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11483768
- Grose C, Buckingham EM, Carpenter JE, Kunkel JP. Varicella-zoster virus infectious cycle: ER stress, autophagic flux, and amphisome-mediated trafficking. Pathogens. 2016; 5: 67. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27973418
- Lenac Roviš T, Bailer SM, Pothineni VR, Ouwendijk WJ, Šimić H, et al. Comprehensive analysis of varicella-zoster virus proteins using a new monoclonal antibody collection. J Virol. 2013; 87: 6943-6954. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23596286
- Fan Y, Sanyal S, Bruzzone R. Breaking bad: how viruses subvert the cell cycle. Front. Cell Infect Microbiol. 2018; 8: 396. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30510918
- Kawai K, Yawn BP. Risk factors for herpes zoster: a systematic review and meta-analysis. Mayo Clin Proc. 2017; 92: 1806-1821. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29202939
- Ivanova L, Tzaneva D, Stoykova Z, Kostadinova T. Viral diseases in transplant and immunocompromised patients. In: Immunopathology and immunomodulation. Intech Open. 2015.
- Pergam SA, Limaye AP. AST Infectious Diseases Community of Practice: Varicella zoster virus (VZV) in solid organ transplant recipients. Am J Transplant. 2009; 9: 108-115. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20070670
- Wiegering V, Schick J, Beer M, Weissbrich B, Gattenlöhner et al. Varicellazoster virus infections in immunocompromised patients - a single centre 6-years analysis. BMC Pediatr. 2011; 11: 31. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21569228
- Folatre I, Zolezzi P, Schmidt D, Marín F, Täger M. Infections caused by Varicella zoster virus in children with cancer aged less than 15 years old. Rev Med Chil. 2003; 131: 759-764. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14513696
- Marra F, Lo E, Kalashnikov V, Richardson K. Risk of herpes zoster in individuals on biologics, disease-modifying antirheumatic drugs and/or corticosteroids for autoimmune diseases: a systematic review and meta-analysis. Open Forum Infect Dis. 2016; 3: 205. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27942537
- Fan L, Wang Y, Liu X, Guan X. Association between statin use and herpes zoster: systematic review and meta-analysis. BMJ Open. 2019; 9: 022897. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30765397
- Schmidt SA, Mor A, Schønheyder HC, Sørensen HT, Dekkers OM, et al. Herpes zoster as a marker of occult cancer: a systematic review and meta-analysis. J Infect. 2017; 74: 215-235. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27845154
- Albrecht MA, Levin MJ. Epidemiology, clinical manifestations, and diagnosis of herpes zoster. Up To Date. 2019.
- Ogunjimi B, Zhang SY, Sørensen KB, Skipper KA, Carter-Timofte M, et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J Clin Invest. 2017; 127: 3543-3556. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28783042
- Wung PK, Holbrook JT, Hoffman GS, Tibbs AK, Specks U, et al. WGET Research Group. Herpes zoster in immune compromised patients: incidence, timing, and risk factors. Am J Med. 2005; 118: 1416.
- Kanbayashi Y, Matsumoto Y, Kuroda J, Kobayashi T, Horiike S, et al. Predicting risk factors for varicella zoster virus infection and postherpetic neuralgia after hematopoietic cell transplantation using ordered logistic regression analysis. Ann Hematol. 2017; 96: 311-315. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27896415
- Leung TF, Chik KW, Li CK, Lai H, Shing MM, et al. Incidence, risk factors and outcome of varicella-zoster virus infection in children after haematopoietic stem cell transplantation. Bone Marrow Transplant. 2000; 25: 167-172. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10673675
- Crosslin DR; Carrell DS, Burt A, Kim DS, Underwood JG, et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015; 16: 1-7. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25297839
- Sandherr M, Hentrich M, von Lilienfeld-Toal M, Massenkeil G, Neumann S, et al. Antiviral prophylaxis in patients with solid tumours and haematological malignancies--update of the Guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society for Hematology and Medical Oncology (DGHO). Ann Hematol. 2015; 94: 1441-1450. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26193852
- Sandherr M, Einsele H, Hebart H, Kahl C, Kern W, et al. Infectious Diseases Working Party, German Society for Hematology and Oncology. Antiviral prophylaxis in patients with haematological malignancies and solid tumours: Guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society for Hematology and Oncology (DGHO). Ann Oncol. 2006; 17: 10511059. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16410361
- Yetgin S, Kuşkonmaz B, Aytaç S, Cetin M. The evaluation of acquired aplastic anemia in children and unexpected frequency of varicella-zoster virus association: a single-center study. Turk J Pediatr. 2008; 50: 342-348. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19014047
- Gulick RM, Heath-Chiozzi M, Crumpacker CS. Varicella-zoster virus disease in patients with human immunodeficiency virus infection. Arch Dermatol. 1990; 126: 1086-1088. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2200349
- Alcaide ML, Abbo L, Pano JR, Gaynor JJ, Tryphonopoulos P, et al. Herpes zoster infection after liver transplantation in patients receiving induction therapy with alemtuzumab. Clin Transplant. 2008; 22: 502-507. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18627401
- Dogra M, Bajgai P, Kumar A, Sharma A. Progressive outer retinal necrosis after rituximab and cyclophosphamide therapy. Indian J Ophthalmol. 2018; 66: 591-593. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29582832
- Tsuji H, Yoshifuji H, Fujii T, Matsuo T, Nakashima R, et al. Visceral disseminated varicella zoster virus infection after rituximab treatment for granulomatosis with polyangiitis. Mod Rheumatol. 2017; 27: 155-161. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25159158
- König C, Kleber M, Reinhardt H, Knop S, Wäsch R, et al. Incidence, risk factors, and implemented prophylaxis of varicella zoster virus infection, including complicated varicella zoster virus and herpes simplex virus infections, in lenalidomide-treated multiple myeloma patients. Ann Hematol. 2014; 93: 479484. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24318541
- Curley MJ, Hussein SA, Hassoun PM. Disseminated herpes simplex virus and varicella zoster virus coinfection in a patient taking thalidomide for relapsed multiple myeloma. J Clin Microbiol. 2002; 40: 2302-2304. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12037117
- Thomas SL, Hall AJ. What does epidemiology tell us about risk factors for herpes zoster? Lancet Infect Dis. 2004; 4: 26-33. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14720565
- Kim JW, Min CK, Mun YC, Park Y, Kim BS, et al. Varicella-zoster virusspecific cell-mediated immunity and herpes zoster development in multiple myeloma patients receiving bortezomib- or thalidomide-based chemotherapy. J Clin Virol. 2015; 73: 64-69. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26546878
- Park H, Youk J, Kim HR, Koh Y, Kwon JH, et al. Infectious complications in multiple myeloma receiving autologous stem cell transplantation in the past 10 years. Int J Hematol. 2017; 106: 801-810. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28825207
- Carter-Timofte ME, Paludan SR, Mogensen TH. RNA polymerase III as a gatekeeper to prevent severe VZV infections. Trends Mol Med. 2018; 24: 904915. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30115567
- Al-Jasser AM, Al-Anazi KA. Infections in patients with multiple myeloma in the era of novel agents and stem cell therapiues. In: Update on multiple myeloma. Intech Open 2018.
- Mazzarello V, Ferrari M, Decandia S, Sotgiu MA. Sunlight and herpes virus. In: Human herpesvirus infection - biological features, transmission, symptoms, diagnosis and treatment. Intech Open 2018.
- Liu J, Wang M, Gan L, Yang S, Chen J. Genotyping of clinical varicella-zoster virus isolates collected in China. J Clin Microbiol. 2009; 47: 1418-1423. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19244468
- Kim KH, Choi YJ, Song KH, Park WB, Jeon JH, et al. Genotype of varicellazoster virus isolates in South Korea. J Clin Microbiol. 2011; 49: 1913-1916. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21411584
- Bostikova V, Salavec M, Smetana J, Chlibek R, Kosina P, et al. Genotyping of varicella-zoster virus (VZV) wild-type strains isolated in the Czech Republic. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011; 155: 379-384. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22336652
- Loparev V, Martro E, Rubtcova E, Rodrigo C, Piette JC, et al. Toward universal varicella-zoster virus (VZV) genotyping: diversity of VZV strains from France and Spain. J Clin Microbiol. 2007; 45: 559-563. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17135433
- Zell R, Taudien S, Pfaff F, Wutzler P, Platzer M, et al. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J Virol. 2012; 86: 1608-1622. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22130537
- Breuer J, Grose C, Norberg P, Tipples G, Schmid DS. A proposal for a common nomenclature for viral clades that form the species varicella-zoster virus: summary of VZV Nomenclature Meeting 2008, Barts and the London School of Medicine and Dentistry. J Gen Virol. 2010; 91: 821-828. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20071486
- Sauerbrei A, Zell R, Philipps A, Wutzler P. Genotypes of varicella-zoster virus wild-type strains in Germany. J Med Virol. 2008; 80: 1123-1130. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18428135
- Loparev VN, Rubtcova EN, Bostik V, Tzaneva V, Sauerbrei A, et al. Distribution of varicella-zoster virus (VZV) wild-type genotypes in Northern and Southern Europe: Evidence for high conservation of circulating genotypes. Virology. 2009; 383: 216-225. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19019403
- Loparev VN, Rubtcova EN, Bostik V, Govil D, Birch CJ, et al. Identification of five major and two minor genotypes of varicella-zoster virus strains: a practical two-amplicon approach used to genotype clinical isolates in Australia and New Zealand. J Virol. 2007; 81: 12758-12765. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17898056
- Toi CS, Dwyer DE. Prevalence of varicella-zoster virus genotypes in Australia characterized by high-resolution melt analysis and ORF22 gene analyses. J Med Microbiol. 2010; 59: 935-940. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20466839
- Buckingham EM, Jarosinski KW, Jackson W, Carpenter JE, Grose C. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016; 90: 8673-8685. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27440906
- Freer G, Pistello M. Varicella-zoster virus infection: natural history, clinical manifestations, immunity and current and future vaccination strategies. New Microbiol. 2018; 41: 95-105. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29498740
- Shaw J, Gershon AA. Varicella virus vaccination in the United States. Viral Immunol. 2018; 31: 96-103. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29173081
- Cohrs RJ, Lee KS, Beach A, Sanford B, Baird NL, et al. Targeted genome sequencing reveals varicella-zoster virus open reading frame 12 deletion. J Virol. 2017; 91. 01141-01147. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28747504
- Gershon M, Gershon A. Varicella-zoster virus and the enteric nervous system. J. Infect. Dis. 2018; 218: 113-119. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30247599
- Ruyechan WT. Roles of cellular transcription factors in VZV replication. Curr Top Microbiol Immunol. 2010; 342: 43-65. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20490778
- Grigoryan S, Yee MB, Glick Y, Gerber D, Kepten E, et al. Direct transfer of viral and cellular proteins from varicella-zoster virus-infected non-neuronal cells to human axons. PLoS One. 2015; 10: 0126081.
- Ambagala AP, Bosma T, Ali MA, Poustovoitov M, Chen JJ, et al. Varicellazoster virus immediate-early 63 protein interacts with human antisilencing function 1 protein and alters its ability to bind histones h3.1 and h3.3. J Virol. 2009; 83: 200-209.
- Silmon de Monerri NC, Kim K. Pathogens hijack the epigenome: a new twist on host-pathogen interactions. Am J Pathol. 2014; 184: 897-911. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24525150
- Narayanan A, Nogueira ML, Ruyechan WT, Kristie TM. Combinatorial transcription of herpes simplex virus and varicella zoster virus immediate early genes is strictly determined by the cellular coactivator HCF-1. J Biol Chem. 2005; 280: 1369-1375. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15522876
- Sadeghipour S, Mathias RA. Herpesviruses hijack host exosomes for viral pathogenesis. Semin Cell Dev Biol. 2017; 67: 91-100. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28456604
- Tavalai N, Stamminger T. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses. 2009; 1: 1240-1264. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21994592
- Reichelt M, Brady J, Arvin AM. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol. 2009; 83: 3904-3918. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19193797
- Yamamoto T, Ali MA, Liu X, Cohen JI. Activation of H2AX and ATM in varicella-zoster virus (VZV)-infected cells is associated with expression of specific VZV genes. Virology. 2014; 453: 52-58. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24606682
- Leisenfelder SA, Moffat JF. Varicella-zoster virus infection of human foreskin fibroblast cells results in atypical cyclin expression and cyclin-dependent kinase activity. J Virol. 2006; 80: 5577-5587. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16699039
- Depledge DP, Ouwendijk WJD, Sadaoka T, Braspenning SE, Mori Y, et al. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat Commun. 2018; 9: 1167. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29563516
- Yang M, Hay J, Ruyechan WT. The DNA element controlling expression of the varicella-zoster virus open reading frame 28 and 29 genes consists of two divergent unidirectional promoters which have a common USF site. J Virol. 2004; 78: 10939-10952. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15452214
- Leisenfelder SA, Kinchington PR, Moffat JF. Cyclin-dependent kinase 1/cyclin B1 phosphorylates varicella-zoster virus IE62 and is incorporated into virions. J Virol. 2008; 82: 12116-12125. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18799590
- Münz C. The autophagic machinery in viral exocytosis. Front Microbiol. 2017; 8: 269. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28270807
- Buckingham EM, Carpenter JE, Jackson W, Zerboni L, Arvin AM, et al. Autophagic flux without a block differentiates varicella-zoster virus infection from herpes simplex virus infection. Proc Natl Acad Sci USA. 2015; 112: 256-261. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25535384
- Buckingham EM, Jarosinski KW, Jackson W, Carpenter JE, Grose C. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016; 90: 8673-8685. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27440906
- Pleet ML, Branscome H, DeMarino C, Pinto DO, Zadeh MA, et al. Autophagy, EVs, and infections: a perfect question for a perfect time. Front Cell Infect Microbiol. 2018; 8: 362. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30406039
- Graybill C, Morgan MJ, Levin MJ, Lee KS. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology. 2018; 522: 220-227. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30053655
- Guise AJ, Budayeva HG, Diner BA, Cristea IM. Histone deacetylases in herpesvirus replication and virus-stimulated host defense. Viruses. 2013; 5: 1607-1632. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23807710
- Adhya D, Basu A. Epigenetic modulation of host: new insights into immune evasion by viruses. J Biosci. 2010; 35: 647-663. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21289446
- Lieberman PM. Epigenetics and genetics of viral latency. Cell Host Microbe. 2016; 19: 619-628. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27173930
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009; 15: 1312-1317. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19855399
- Van Opdenbosch N, Favoreel H, Van de Walle GR. Histone modifications in herpesvirus infections. Biol Cell. 2012; 104: 139-164.
- Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, et al. Snapshots: chromatin control of viral infection. Virology. 2013; 435: 141-156. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23217624
- Wang Z, Deng Z, Tutton S, Lieberman PM. The telomeric response to viral infection. Viruses. 2017; 9: 218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28792463
- Nehme Z, Pasquereau S, Herbein G. Control of viral infections by epigenetictargeted therapy. Clin Epigenetics. 2019; 11: 55. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30917875
- Onozawa M, Hashino S, Takahata M, Fujisawa F, Kawamura T, et al. Relationship between preexisting anti-varicella-zoster virus (VZV) antibody and clinical VZV reactivation in hematopoietic stem cell transplantation recipients. J Clin Microbiol. 2006; 44: 4441-4443. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17035500
- Cvjetković D, Jovanović J, Hrnjaković-Cvjetković I, Brkić S, Bogdanović M. Reactivation of herpes zoster infection by varicella-zoster virus. Med Pregl. 1999; 52: 125-128. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10518396
- Erskine N, Tran H, Levin L, Ulbricht C, Fingeroth J, et al. A systematic review and meta-analysis on herpes zoster and the risk of cardiac and cerebrovascular events. PLoS One. 2017; 12: 0181565. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28749981
- Nagel MA, Gilden D. Complications of varicella zoster virus reactivation. Curr Treat Options Neurol. 2013; 15: 439-453. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24792344
- Mueller NH, Gilden DH, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus infection: clinical features, molecular pathogenesis of disease, and latency. Neurol. Clin. 2008; 26: 675-697. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18657721
- Gilden D, Nagel MA, Cohrs RJ, Mahalingam R. The variegate neurological manifestations of varicella zoster virus infection. Curr Neurol Neurosci Rep. 2013; 13: 374. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23884722
- Kawai K, Gebremeskel BG, Acosta CJ. Systematic review of incidence and complications of herpes zoster: towards a global perspective. BMJ Open. 2014; 4: 004833. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24916088
- Kalogeropoulos CD, Bassukas ID, Moschos MM, Tabbara KF. Eye and periocular skin involvement in herpes zoster infection. Med Hypothesis Discov. Innov Ophthalmol. 2015; 4: 142-156. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27800502
- Albrecht MA. Diagnosis of varicella zoster virus infection. Up To Date. 2019.
- Sauerbrei A, Taut J, Zell R, Wutzler P. Resistance testing of clinical varicellazoster virus strains. Antiviral Res. 2011; 90: 242-247. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21539861
- Hoffmann A, Döring K, Seeger NT, Bühler M, Schacke M, et al. Genetic polymorphism of thymidine kinase (TK) and DNA polymerase (pol) of clinical varicella-zoster virus (VZV) isolates collected over three decades. J Clin Virol. 2017; 95: 61-65. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28886462
- Morfin F, Thouvenot D, De Turenne-Tessier M, Lina B, Aymard M, et al. Phenotypic and genetic characterization of thymidine kinase from clinical strains of varicella-zoster virus resistant to acyclovir. Antimicrob Agents Chemother. 1999; 43: 2412-2416. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10508017
- Mercier-Darty M, Boutolleau D, Lepeule R, Rodriguez C, Burrel S. Utility of ultra-deep sequencing for detection of varicella-zoster virus antiviral resistance mutations. Antiviral Res. 2018; 151: 20-23. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29337163
- De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev. 2016; 29: 695-747. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27281742
- Andrei G, van den Oord J, Fiten P, Opdenakker G, De Wolf-Peeters C, et al. Organotypic epithelial raft cultures as a model for evaluating compounds against alphaherpesviruses. Antimicrob Agents Chemother. 2005; 49: 4671-4680. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16251311
- Mustafa MB, Arduino PG, Porter SR. Varicella zoster virus: review of its management. J Oral Pathol Med. 2009; 38: 673-688. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19691461
- Breuer J, Whitley R. Varicella zoster virus: natural history and current therapies of varicella and herpes zoster. Herpes. 2007; 14: 25-29. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17939892
- Mullane KM, Nuss C, Ridgeway J, Prichard MN, Hartline CB, et al. Brincidofovir treatment of acyclovir-resistant disseminated varicella zoster virus infection in an immunocompromised host. Transpl Infect Dis. 2016; 18: 785790. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27481400
- Stryjewski TP, Scott NL, Barshak MB, Tobin EH, Mali JO, et al. Treatment of refractory acute retinal necrosis with intravenous foscarnet or cidofovir. Ocul Immunol Inflamm. 2018; 26: 199-203. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27598973
- Wong R, Pavesio CE, Laidlaw DA, Williamson TH, Graham EM, et al. Acute retinal necrosis: the effects of intravitreal foscarnet and virus type on outcome. Ophthalmology. 2010; 117: 556-560. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20031221
- Schoenberger SD, Kim SJ, Thorne JE, Mruthyunjaya P, Yeh S, et al. Diagnosis and treatment of acute retinal necrosis: a report by the American academy of ophthalmology. Ophthalmology. 2017; 124: 382-392. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28094044
- Inoue N, Matsushita M, Fukui Y, Yamada S, Tsuda M, et al. Identification of a varicella-zoster virus replication inhibitor that blocks capsid assembly by interacting with the floor domain of the major capsid protein. J Virol. 2012; 86: 12198-12207. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22933294
- McGuigan C, Pathirana RN, Migliore M, Adak R, Luoni G, et al. Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus. J Antimicrob Chemother. 2007; 60: 1316-1330. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17956908
- McGuigan, C.; Balzarini, J. Aryl furano pyrimidines: the most potent and selective anti-VZV agents reported to date. Antiviral. Res. 2006; 71: 149-153. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16712966
- Balzarini J, McGuigan C. Chemotherapy of varicella-zoster virus by a novel class of highly specific anti-VZV bicyclic pyrimidine nucleosides. Biochim Biophys Acta. 2002; 1587: 287-295. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12084470
- Balzarini J, McGuigan C. Bicyclic pyrimidine nucleoside analogues (BCNAs) as highly selective and potent inhibitors of varicella-zoster virus replication. J Antimicrob Chemother. 2002; 50: 5-9. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12096000
- Migliore M. FV-100: the most potent and selective anti-varicella zoster virus agent reported to date. Antivir Chem Chemother. 2010; 20: 107-115. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20054098
- Kim S.R, Khan F, Ramirez-Fort MK, Downing C, Tyring SK. Varicella zoster: an update on current treatment options and future perspectives. Expert Opin Pharmacother. 2014; 15: 61-71. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24289750
- Johnson RW. Herpes zoster in the immunocompetent patient: management of post-herpetic neuralgia. Herpes. 2003; 10: 38-45. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14577953
- Wang L, Zhu L, Zhu H. Efficacy of varicella (VZV) vaccination: an update for the clinician. Ther Adv Vaccines. 2016; 4: 20-31. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27551429
- Haberthur K, Messaoudi I. Animal models of varicella zoster virus infection. Pathogens. 2013; 2: 364-382. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25437040
- Harada K, Heaton H, Chen J, Vazquez M, Meyer J. Zoster vaccine-associated primary varicella infection in an immunocompetent host. BMJ Case Rep. 2017; 2017: PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28830902
- Gabutti G, Bonanni P, Conversano M, Fanelli G, Franco E, et al. Prevention of herpes zoster and its complications: from clinical evidence to real life experience. Hum Vaccin Immunother. 2017; 13: 391-398. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27925894
- Warren-Gash C, Forbes H, Breuer J. Varicella and herpes zoster vaccine development: lessons learned. Expert Rev. Vaccines. 2017; 16: 1191-1201. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29047317
- Papaloukas O, Giannouli G, Papaevangelou V. Successes and challenges in varicella vaccine. Ther Adv Vaccines. 2014; 2: 39-55. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24757524
- Arnold N, Messaoudi I. Herpes zoster and the search for an effective vaccine. Clin Exp Immunol. 2017; 187: 82-92. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27164323
- Willis ED, Woodward M, Brown E, Popmihajlov Z, Saddier P, et al. Herpes zoster vaccine live: a 10 year review of post-marketing safety experience. Vaccine. 2017; 35: 7231-7239. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29174682
- Leung J, Broder KR, Marin M. Severe varicella in persons vaccinated with varicella vaccine (breakthrough varicella): a systematic literature review. Expert Rev Vaccines. 2017; 16: 391-400. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28276305
- Khandelwal P, Marsh RA, Scott Scmid D, Radford KW, Bleesing J, et al. Case series of vaccine associated varicella zoster virus infection in immune compromised patients. Biol Blood Marrow Transplant. 2013; 19: 250.
- Bhalla P, Forrest GN, Gershon M, Zhou Y, Chen J, et al. Disseminated, persistent, and fatal infection due to the vaccine strain of varicella-zoster virus in an adult following stem cell transplantation. Clin Infect Dis. 2015; 60: 10681074. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25452596
- Wang L, Verschuuren EAM, van Leer-Buter CC, Bakker SJL, de Joode AAE, et al. Herpes zoster and immunogenicity and safety of zoster vaccines in transplant patients: a narrative review of the literature. Front Immunol. 2018; 9: 1632. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30079064
- Aoki T, Koh K, Kawano Y, Mori M, Arakawa Y, et al. Safety of live attenuated high-titer varicella-zoster virus vaccine in pediatric allogeneic hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant. 2016; 22: 771-775. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26748161
- Chou JF, Kernan NA, Prockop S, Heller G, Scaradavou A, et al. Safety and immunogenicity of the live attenuated varicella vaccine following T replete or T cell-depleted related and unrelated allogeneic hematopoietic cell transplantation (alloHCT). Biol Blood Marrow Transplant. 2011; 17: 1708-1713. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21664979
- Winston DJ, Mullane KM, Cornely OA, Boeckh MJ, Brown JW, et al. V212 Protocol 001 Trial Team. Inactivated varicella zoster vaccine in autologous haemopoietic stem-cell transplant recipients: An international, multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2018; 391: 2116-2127. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29856344
- Stadtmauer EA, Sullivan K, Marty F, Dadwal SS, Papanicolaou GA, et al. One-year safety and immunogenicity of two formulations of an adjuvanted varicella-zoster virus (VZV) subunit candidate vaccine in adult autologous hematopoietic cell transplant (HCT) recipients. Biol Blood Marrow Transplant. 2013; 19: 168-169.
- Issa NC, Marty FM, Leblebjian H, Galar A, Shea MM, et al. Live attenuated varicella-zoster vaccine in hematopoietic stem cell transplantation recipients. Biol. Blood Marrow Transplant. 2014; 20: 285-287. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24269706
- Sasadeusz J, Prince HM, Schwarer A, Szer J, Stork A, et al. Immunogenicity and safety of a two-dose live attenuated varicella vaccine given to adults following autologous hematopoietic stem cell transplantation. Transpl Infect Dis. 2014; 16: 1024-1031. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25272081
- Parrino J, McNeil SA, Lawrence SJ, Kimby E, Pagnoni MF, et al. Safety and immunogenicity of inactivated varicella-zoster virus vaccine in adults with hematologic malignancies receiving treatment with anti-CD20 monoclonal antibodies. Vaccine. 2017; 35: 1764-1769. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28268074
- Leung TF, Li CK, Hung EC, Chan PK, Mo CW, et al. Immunogenicity of a two-dose regime of varicella vaccine in children with cancers. Eur J Haematol. 2004; 72: 353-357. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15059071
- Ohfuji S, Ito K, Inoue M, Ishibashi M, Kumashiro H, et al. Safety of live attenuated varicella-zoster vaccine in patients with underlying illnesses compared with healthy adults: a prospective cohort study. BMC Infect Dis. 2019; 19: 95. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30691396
- Eberhardson M, Hall S, Papp KA, Sterling TM, Stek JE, et al. Safety and immunogenicity of inactivated varicella-zoster virus vaccine in adults with autoimmune disease: a phase 2, randomized, double-blind, placebo-controlled clinical trial. Clin Infect Dis. 2017; 65: 1174-1182. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29126292
- Russell AF, Parrino J, Fisher CL Jr, Spieler W, Stek JE, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects on chronic/maintenance corticosteroids. Vaccine. 2015; 33: 3129-3134. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25964168
- Senderovich H, Grewal J, Mujtaba M. Herpes zoster vaccination efficacy in the long-term care facility population: a qualitative systematic review. Curr Med Res Opin. 2019: 1-12. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30913912
- Mills R, Tyring SK, Levin MJ, Parrino J, Li X, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010; 28: 4204-4209. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20416263
- Santos KB, Souza RS, Atalla A, Hallack-Neto AE. Herpes zoster after autologous hematopoietic stem cell transplantation. Rev Bras Hematol Hemoter. 2016; 38: 298-301. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27863756
- Truong Q, Veltri L, Kanate AS, Hu Y, Craig M, et al. Impact of the duration of antiviral prophylaxis on rates of varicella-zoster virus reactivation disease in autologous hematopoietic cell transplantation recipients. Ann Hematol. 2014; 93: 677-682. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24097085
- Wada-Shimosato Y, Tanoshima R, Hiratoko K, Takeuchi M, Tsujimoto SI, et al. Effectiveness of acyclovir prophylaxis against varicella zoster virus disease after allogeneic hematopoietic cell transplantation: a systematic review and metaanalysis. Transpl Infect Dis. 2019; 13061.
- Okuma HS, Kobayashi Y, Makita S, Kitahara H, Fukuhara S, et al. Disseminated herpes zoster infection initially presenting with abdominal pain in patients with lymphoma undergoing conventional chemotherapy: a report of three cases. Oncol Lett. 2016; 12: 809-814. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27446355
- Sahoo F, Hill JA, Xie H, Leisenring W, Yi J, Goyal S, et al. Herpes zoster in autologous hematopoietic cell transplant recipients in the era of acyclovir or valacyclovir prophylaxis and novel treatment and maintenance therapies. Biol Blood Marrow Transplant. 2017; 23: 505-511. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28039754
- Kim SK, Kim MC, Han SB, Kim SK, Lee JW, et al. Clinical characteristics and outcomes of varicella zoster virus infection in children with hematologic malignancies in the acyclovir era. Blood Res. 2016; 51: 249-255. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28090487
- Kanda Y, Mineishi S, Saito T, Saito A, Yamada S, et al. Long-term low-dose acyclovir against varicella-zoster virus reactivation after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001; 28: 689-692. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11704792
- Asano-Mori Y, Kanda Y, Oshima K, Kako S, Shinohara A, et al. Long-term ultra-low-dose acyclovir against varicella-zoster virus reactivation after allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2008; 83: 472-476. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18266207
- Han SB, Kim SK, Lee JW, Lee DG, Chung NG, et al. Varicella zoster virus infection after allogeneic hematopoietic cell transplantation in children using a relatively short duration of acyclovir prophylaxis: a retrospective study. Medicine. 2017; 96: 6546. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28383421
- Goodwin TJ, McCarthy M, Osterrieder N, Cohrs RJ, Kaufer BB. Three dimensional normal human neural progenitor tissue-like assemblies: a model of persistent varicella-zoster virus infection. PLoS Pathog. 2013; 9: 1003512. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23935496
- Shahzad A, Gilden D, Cohrs RJ. Translational medicine and varicella zoster virus: need for disease modeling. New Horiz Transl Med. 2015; 2: 89-91. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26086038
- Messaoudi I, Barron A, Wellish M, Engelmann F, Legasse A, et al. Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog. 2009; 5: 1000657. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19911054
- Steain M, Slobedman B, Abendroth A. Experimental models to study varicella-zoster virus infection of neurons. Curr Top Microbiol Immunol. 2010; 342: 211-228. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20373093
- White TM, Gilden DH, Mahalingam R. An animal model of varicella virus infection. Brain Pathol. 2001; 11: 475-479. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11556693
- Moffat JF, Zerboni L, Sommer MH, Heineman TC, Cohen JI, et al. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc Natl Acad Sci USA. 1998; 95: 11969-11974. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9751774
- Zerboni L, Sung P, Sommer M, Arvin A. The C-terminus of varicella-zoster virus glycoprotein M contains trafficking motifs that mediate skin virulence in the SCID-human model of VZV pathogenesis. Virology. 2018; 523: 110-120. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30119012
- Lee KS, Zhou W, Scott-McKean JJ, Emmerling KL, Cai GY, et al. Human sensory neurons derived from induced pluripotent stem cells support varicellazoster virus infection. PLoS One. 2012; 7: 53010. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23285249
- Markus A, Lebenthal-Loinger I, Yang IH, Kinchington PR, Goldstein RS. An in vitro model of latency and reactivation of varicella zoster virus in human stem cell-derived neurons. PLoS Pathog. 2015; 11: 1004885. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26042814
- Sadaoka T, Schwartz CL, Rajbhandari L, Venkatesan A, Cohen JI. Human embryonic stem cell-derived neurons are highly permissive for varicella-zoster virus lytic infection. J Virol. 2017; 92: 01108-01117. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29046461
- Dukhovny A, Sloutskin A, Markus A, Yee MB, Kinchington PR, et al. Varicella-zoster virus infects human embryonic stem cell-derived neurons and neurospheres but not pluripotent embryonic stem cells or early progenitors. J Virol. 2012; 86: 3211-3218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22238301
- Sloutskin A, Kinchington PR, Goldstein RS. Productive vs non-productive infection by cell-free varicella zoster virus of human neurons derived from embryonic stem cells is dependent upon infectious viral dose. Virology. 2013; 443: 285-293. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23769240
- Birenboim R, Markus A, Goldstein RS. Simple generation of neurons from human embryonic stem cells using agarose multiwell dishes. J Neurosci Methods. 2013; 214: 9-14. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23313848
- Baird NL, Zhu S, Pearce CM, Viejo-Borbolla A. Current in vitro models to study varicella zoster virus latency and reactivation. Viruses. 2019; 11: 103. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30691086
- Como CN, Pearce CM, Cohrs RJ, Baird NL. Interleukin-6 and type 1 interferons inhibit varicella zoster virus replication in human neurons. Virology. 2018; 522: 13-18. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29979960
- Baird NL, Bowlin JL, Hotz TJ, Cohrs RJ, Gilden D. Interferon gamma prolongs survival of varicella-zoster virus-infected human neurons in vitro. J Virol. 2015; 89: 7425-7427. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25948748
- Li X, Li X, Gong W, Wang G, Lu Z, Wu N, et al. Titration of cell-associated varicella-zoster virus with the MV9G reporter cell line for antiviral studies. J Virol Methods. 2018; 260: 14-20. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29966597
- Wang GQ, Suzutani T, Yamamoto Y, Fukui Y, Nozawa N, et al. Generation of a reporter cell line for detection of infectious varicella-zoster virus and its application to antiviral studies. Antimicrob Agents Chemother. 2006; 50: 3142-3145. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16940113
- Sloutskin A, Goldstein RS. Laboratory preparation of varicella-zoster virus: concentration of virus-containing supernatant, use of a debris fraction and magnetofection for consistent cell-free VZV infections. J Virol Methods. 2014; 206: 128-132. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24925132
- Kumar R, Godavarthy PS, Krause DS. The bone marrow microenvironment in health and disease at a glance. J Cell Sci. 2018; 131: 201707. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29472498
- Ghazanfari B, Behrava J. An update on the components and functions of bone marrow niche. Ann Hematol Oncol. 2017; 4: 1178.
- Chitteti BR, Cheng YH, Poteat B, Rodriguez-Rodriguez S, Goebel WS, et al. Impact of interactions of cellular components of the bone marrow microenvironment on hematopoietic stem and progenitor cell function. Blood. 2010; 115: 3239-3248. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20154218
- Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014; 35:32-37. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24210164
- Huang X, Zhu B, Wang X, Xiao R, Wang C. Three-dimensional co-culture of mesenchymal stromal cells and differentiated osteoblasts on human bio-derived bone scaffolds supports active multi-lineage hematopoiesis in vitro: Functional implication of the biomimetic HSC niche. Int J Mol Med. 2016; 38: 1141-1151. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27571775
- Isern J, García-García A, Martín AM, Arranz L, Martín-Pérez D, et al. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. Elife. 2014; 3: 03696. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25255216
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, et al. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. 2013; 210: 1351-1367. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23776077
- Lucas D. The bone marrow microenvironment for hematopoietic stem cells. Adv Exp Med Biol. 2017; 1041: 5-18.
- Eltoukhy HS, Sinha G, Moore CA, Gergues M, Rameshwar P. Secretome within the bone marrow microenvironment: a basis for mesenchymal stem cell treatment and role in cancer dormancy. Biochimie. 2018; 155: 92-103. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29859990
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stemcell maintenance. Nature. 2013; 495: 227-230. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23434756
- Agarwala S, Tamplin OJ. Neural crossroads in the hematopoietic stem cell niche. Trends Cell Biol. 2018; 28: 987-998. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29857963
- Coste C, Neirinckx V, Gothot A, Wislet S, Rogister B. Are neural crest stem cells the missing link between hematopoietic and neurogenic niches? Front Cell Neurosci. 2015; 9: 218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26136659
- Lotem J, Sachs L. Cytokine control of developmental programs in normal hematopoiesis and leukemia. Oncogene. 2002; 21: 3284-3294. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12032770
- Ackermann M, Liebhaber S, Klusmann JH, Lachmann N. Lost in translation: pluripotent stem cell-derived hematopoiesis. EMBO Mol Med. 2015; 7: 13881402. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26174486
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008; 132: 631-644. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18295580
- Mirantes C, Passegué E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp. Cell. Res. 2014; 329: 248-254. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25149680
- Glatman Zaretsky A, Engiles JB, Hunter CA. Infection-induced changes in hematopoiesis. J Immunol. 2014; 192: 27-33. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24363432
- Metcalf D. Hematopoietic cytokines. Blood. 2008; 111: 485-491.
- Boiko JR, Borghesi L. Hematopoiesis sculpted by pathogens: Toll-like receptors and inflammatory mediators directly activate stem cells. Cytokine. 2012; 57: 1-8. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22079335
- Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol. 2008; 15: 307-311. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18536567
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015; 163: 1663-1677. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26627738
- Brown G, Ceredig R, Tsapogas P. The making of hematopoiesis: developmental ancestry and environmental nurture. Int J Mol Sci. 2018; 19: 2122. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30037064
- Zhao JL, Baltimore D. Regulation of stress-induced hematopoiesis. Curr Opin Hematol. 2015; 22: 286-292. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26049748
- Schuettpelz LG, Link DC. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol. 2013; 4: 204. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23882270
- Croker BA, Silke J, Gerlic M. Fight or flight: regulation of emergency hematopoiesis by pyroptosis and necroptosis. Curr Opin Hematol. 2015; 22: 293-301. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26049749
- Pascutti MF, Erkelens MN, Nolte MA. Impact of viral infections on hematopoiesis: from beneficial to detrimental effects on bone marrow output. Front Immunol. 2016; 7: 364. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27695457
- Ardalan MR, Shoja MM, Tubbs RS, Esmaili H, Keyvani H. Postrenal transplant hemophagocytic lymphohistiocytosis and thrombotic microangiopathy associated with parvovirus B19 infection. Am J Transplant. 2008; 8: 1340-1344. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18522549
- Simmons P, Kaushansky K, Torok-Storb B. Mechanisms of cytomegalovirusmediated myelosuppression: perturbation of stromal cell function versus direct infection of myeloid cells. Proc Natl Acad Sci USA. 1990; 87: 1386-1390. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2154745
- Al-Uzri A, Yorgin PD, Kling PJ. Anemia in children after transplantation: etiology and the effect of immunosuppressive therapy on erythropoiesis. Pediatr Transplant. 2003; 7: 253-264. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12890002
- Torok-Storb B, Simmons P, Khaira D, Stachel D, Myerson D. Cytomegalovirus and marrow function. Ann Hematol. 1992; 64: 128-131.
- Randolph-Habecker J, Iwata M, Torok-Storb B. Cytomegalovirus mediated myelosuppression. J Clin Virol. 2002; 25: 51-56. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12361756
- Movassagh M, Gozlan J, Senechal B, Baillou C, Petit JC, et al. Direct infection of CD34+ progenitor cells by human cytomegalovirus: evidence for inhibition of hematopoiesis and viral replication. Blood. 1996; 88: 1277-1283. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8695845
- Sing GK, Ruscetti FW. The role of human cytomegalovirus in haematological diseases. Baillieres Clin Haematol. 1995; 8: 149-163. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7663045
- Kakish K, Basak RB, Al Dhuhouri J, Chakraborty S. Four year old child with breakthrough varicella leading to pancytopenia. Bahrain Med Bull. 2009; 31: 2.
- Muthu V, Kumar PS, Varma S, Malhotra P. Varicella zoster virus-related pancytopenia. Int J Infect Dis. 2013; 17: 1264. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23911240
- Kuskonmaz B, Cetin M, Uckan D, Yetgin S. Varicella zoster-associated severe aplastic anemia in a child and its successful treatment with peripheral blood stem cell transplantation from HLA-5/6-identical donor. Med Sci Monit. 2007; 13: 128-131. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17901857
- Ragozzino MW, Melton LJ. Kurland LT, Chu CP, Perry HO. Risk of cancer after herpes zoster: a population-based study. N Engl J Med. 1982; 307: 393-397. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/6979711
- Ho JD, Xirasagar S, Lin HC. Increased risk of a cancer diagnosis after herpes zoster ophthalmicus: a nationwide population-based study. Ophthalmology. 2011; 118: 1076-1081. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21232800
- Schmidt SA, Mor A, Schønheyder HC, Sørensen HT, Dekkers OM, et al. Herpes zoster as a marker of occult cancer: A systematic review and meta-analysis. J Infect. 2017; 74: 215-235. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27845154
- Liu YC, Yang YH, Hsiao HH, Yang WC, Liu TC, et al. Herpes zoster is associated with an increased risk of subsequent lymphoid malignancies - a nationwide population-based matched-control study in Taiwan. BMC Cancer. 2012; 12: 503. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23114019
- Mays RM, Murthy RK, Gordon RA, Lapolla WJ, Galfione SK, et al. Diffuse Large B-Cell Lymphoma at the site of a herpes zoster scar. World J Oncol. 2012; 3: 199-203. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29147306
- Thanunchai M, Hongeng S, Thitithanyanont A. Mesenchymal stromal cells and viral infection. Stem Cells Int. 2015; 2015: 860950. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26294919
- Yang K, Wang J, Wu M, Li M, Wang Y, et al. Mesenchymal stem cells detect and defend against gammaherpesvirus infection via the cGAS-STING pathway. Sci. Rep. 2015; 5: 7820. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25592282
- Al-Anazi KA, Al-Jasser AM. Mesenchymal stem cells-their antimicrobial effects and their promising future role as novel therapies of infectious complications in high risk patients. In: Progress in stem cell transplantation. Intech Open. 2015.
- Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. 2014; 6: 552-570. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25426252
- Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, et al. Role for interferongamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006; 24: 386-398. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16123384
- Auletta JJ, Deans RJ, Bartholomew AM. Emerging roles for multipotent, bone marrow-derived stromal cells in host defense. Blood. 2012; 119: 18011809. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22228625
- Boomsma RA, Geenen DL. Mesenchymal stem cells secrete multiple cytokines that promote angiogenesis and have contrasting effects on chemotaxis and apoptosis. PLoS One. 2012; 7: 35685. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22558198
- Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010; 466: 829-834. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20703299
- Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptorexpressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014; 15: 154-168. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24953181
- Pleyer L, Valent P, Greil R. Mesenchymal stem and progenitor cells in normal and dysplastic hematopoiesis-masters of survival and clonality? Int J Mol Sci. 2016; 17: 1009. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27355944
- Shi C. Recent progress toward understanding the physiological function of bone marrow mesenchymal stem cells. Immunology. 2012; 136: 133-138. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22321024
- Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009; 4: 206-216. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19265660
- Liu ZJ, Zhuge Y, Velazquez OC. Trafficking and differentiation of mesenchymal stem cells. J Cell Biochem. 2009; 106: 984-991. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19229871
- De Luca L, Trino S, Laurenzana I, Lamorte D, Caivano A, et al. Mesenchymal stem cell derived extracellular vesicles: a role in hematopoietic transplantation? Int J Mol Sci. 2017; 18: 1022. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28486431
- Castro-Manrreza ME, Montesinos JJ. Immunoregulation by mesenchymal stem cells: biological aspects and clinical applications. J Immunol Res. 2015; 2015: 394917. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25961059
- Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, et al. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3dioxygenase-mediated tryptophan degradation. Blood. 2004; 103: 4619-4621. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15001472
- Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003; 75: 389-397. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12589164
- Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E (2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009; 15: 42-49. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19098906
- Abarbanell AM, Coffey AC, Fehrenbacher JW, Beckman DJ, Herrmann JL, et al. Proinflammatory cytokine effects on mesenchymal stem cell therapy for the ischemic heart. Ann Thorac Surg. 2009; 88: 1036-1043. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19699961
- Leuning DG, Beijer NRM, du Fossé NA, Vermeulen S, Lievers E, et al. The cytokine secretion profile of mesenchymal stromal cells is determined by surface structure of the microenvironment. Sci Rep. 2018; 8: 7716. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29769543
- Rasmusson I, Le Blanc K, Sundberg B, Ringdén O. Mesenchymal stem cells stimulate antibody secretion in human B cells. Scand J Immunol. 2007; 65: 336-343. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17386024
- Avanzi S, Leoni V, Rotola A, Alviano F, Solimando L, et al. Susceptibility of human placenta derived mesenchymal stromal/stem cells to human herpesviruses infection. PLoS One. 2013; 8: 71412. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23940750
- Hersh DS, Wadajkar AS, Roberts N, Perez JG, Connolly NP, et al. Evolving drug delivery strategies to overcome the blood brain barrier. Curr Pharm Des. 2016; 22: 1177-1193. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26685681
- Abdi Z, Eskandary H, Nematollahi-Mahani SN. Effects of two types of human cells on outgrowth of human glioma in rats. Turk Neurosurg. 2018; 28: 19-28. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27943226
- Dong HJ, Li G, Meng HP, Shang CZ, Luo Y, et al. How can mesenchymal stem cells penetrate the blood brain barrier? Turk Neurosurg. 2018; 28: 10131014. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29569697
- Conaty P, Sherman LS, Naaldijk Y, Ulrich H, Stolzing A, et al. Methods of mesenchymal stem cell homing to the blood-brain barrier. Methods Mol Biol. 2018; 1842: 81-91. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30196403
- Liu L, Eckert MA, Riazifar H, Kang DK, Agalliu D, et al. From blood to the brain: can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013; 2013: 435093. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23997771
- Christodoulou I, Goulielmaki M, Devetzi M, Panagiotidis M, Koliakos G, et al. Mesenchymal stem cells in preclinical cancer cytotherapy: a systematic review. Stem Cell Res Ther. 2018; 9: 336. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30526687
- Lee HY, Hong IS. Double-edged sword of mesenchymal stem cells: Cancerpromoting versus therapeutic potential. Cancer Sci. 2017; 108: 1939-1946. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28756624
- Nowakowski A, Drela K, Rozycka J, Janowski M, Lukomska B. Engineered mesenchymal stem cells as an anti-cancer Trojan horse. Stem Cells Dev. 2016; 25: 1513-1531. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27460260
- Zhang L, Su XS, Ye JS, Wang YY, Guan Z, et al. Bone marrow mesenchymal stem cells suppress metastatic tumor development in mouse by modulating immune system. Stem Cell Res Ther. 2015; 6: 45. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25889932
- Fakiruddin KS, Ghazalli N, Lim MN, Zakaria Z, Abdullah S. Mesenchymal stem cell expressing TRAIL as targeted therapy against sensitised tumor. Int J Mol Sci. 2018; 19: 2188. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30060445
- Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018; 154: 3-20. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29313948
- Abendroth A, Morrow G, Cunningham AL, Slobedman B. Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J Virol. 2001; 75: 6183-6192. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11390620
- Morrow G, Slobedman B, Cunningham AL, Abendroth A. Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J Virol. 2003; 77: 4950-4959. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12663800
- Sloan E, Henriquez R, Kinchington PR, Slobedman B, Abendroth A. Varicella-zoster virus inhibition of the NF-κB pathway during infection of human dendritic cells: role for open reading frame 61 as a modulator of NF-κB activity. J Virol. 2012; 86: 1193-202. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22090112
- Hu H, Cohen JI. Varicella-zoster virus open reading frame 47 (ORF47) protein is critical for virus replication in dendritic cells and for spread to other cells. Virology. 2005; 337: 304-311. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15913699
- Pollara G, Kwan A, Newton PJ, Handley ME, Chain BM, et al. Dendritic cells in viral pathogenesis: protective or defective? Int J Exp Pathol. 2005; 86: 187-204. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16045541
- Schönrich G, Raftery MJ. Dendritic cells as Achilles' heel and Trojan horse during varicella zoster virus infection. Front Microbiol. 2015; 6: 417. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26005438
- Huch JH, Cunningham AL, Arvin AM, Nasr N, Santegoets SJ, et al. Impact of varicella-zoster virus on dendritic cell subsets in human skin during natural infection. J Virol. 2010; 84: 4060-4072. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20130046
- Collins PL, Cella M, Porter SI, Li S, Gurewitz GL, Hong HS, et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. 2019; 176: 348-360. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30595449
- Yoon SR, Kim TD, Choi I. Understanding of molecular mechanisms in natural killer cell therapy. Exp Mol Med. 2015; 47: 141. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25676064
- Mehta RS, Randolph B, Daher M, Rezvani K. NK cell therapy for hematologic malignancies. Int J Hematol. 2018; 107: 262-270. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29383623
- Handgretinger R, Lang P, André MC. Exploitation of natural killer cells for the treatment of acute leukemia. Blood. 2016; 127: 3341-3349. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27207791
- See DM, Khemka P, Sahl L, Bui T, Tilles JG. The role of natural killer cells in viral infections. Scand J Immunol. 1997; 46: 217-224. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9315107
- 288 Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, et al. High dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 2018; 49: 971-986. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30413361
- Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017; 47: 820-833. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29166586
- Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010; 142: 847-856. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20850008
- Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018; 9: 1869. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30150991
- Van Erp EA, van Kampen MR, van Kasteren PB, de Wit J. Viral infection of human natural killer cells. Viruses. 2019; 11: 243. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27160662
- Tesi B, Schlums H, Cichocki F, Bryceson YT. Epigenetic regulation of adaptive NK cell diversification. Trends Immunol. 2016; 37: 451-461. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27160662
- Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell. 2018; 172: 534548. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29275861
- Dyck L, Lynch L. New job for NK cells: architects of the tumor microenvironment. Immunity. 2018; 48: 9-11. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29343443
- Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018; 172: 1022-1037. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29429633
- Chouaib S, Pittari G, Nanbakhsh A, El Ayoubi H, Amsellem S, et al. Improving the outcome of leukemia by natural killer cell-based immunotherapeutic strategies. Front Immunol. 2014; 5: 95. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24672522
- Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018; 9: 283. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29497427
- Cooley S, Parham P, Miller JS. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood. 2018; 131: 1053-1062. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29358179
- Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002; 295: 2097-2100. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11896281
- Waggoner SN, Reighard SD, Gyurova IE, Cranert SA, Mahl SE, et al. Roles of natural killer cells in antiviral immunity. Curr Opin Virol. 2016; 16: 15-23. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26590692
- Hammer Q, Romagnani C. About training and memory: NK-cell adaptation to viral infections. Adv Immunol. 2017; 133: 171-207. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28215279
- Campbell TM, McSharry BP, Steain M, Ashhurst TM, Slobedman B, et al. Varicella zoster virus productively infects human natural killer cells and manipulates phenotype. PLoS Pathog. 2018; 14: e1006999. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29709039
- Weinberg A, Levin MJ. VZV T cell-mediated immunity. Curr Top Microbiol Immunol. 2010; 342: 341-357. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20473790
- Frey CR, Sharp MA, Min AS, Schmid DS, Loparev V, et al. Identification of CD8+ T cell epitopes in the immediate early 62 protein (IE62) of varicella-zoster virus, and evaluation of frequency of CD8+ T cell response to IE62, by use of IE62 peptides after varicella vaccination. J Infect Dis. 2003; 188: 40-52. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12825169
- Kim AR, Park J, Kim JH, Kwak JE, Cho Y, et al. Herpes zoster DNA vaccines with IL-7 and IL-33 molecular adjuvants elicit protective T cell immunity. Immune Netw. 2018; 18: e38. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30402333
- van Besouw NM, Verjans GM, Zuijderwijk JM, Litjens NH, Osterhaus AD, et al. Systemic varicella zoster virus reactive effector memory T-cells impaired in the elderly and in kidney transplant recipients. J Med Virol. 2012; 84: 2018-2025. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23080511
- Qi Q, Cavanagh MM, Le Saux S, Wagar LE, Mackey S, et al. Defective T memory cell differentiation after varicella zoster vaccination in older individuals. PLoS Pathog. 2016; 12: e1005892. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27764254
- Vukmanovic-Stejic M, Sandhu D, Seidel JA, Patel N, Sobande TO, et al. The characterization of varicella zoster virus-specific T cells in skin and blood during aging. J Invest Dermatol. 2015; 135: 1752-1762. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25734814
- Arvin AM. Cell-mediated immunity to varicella-zoster virus. J Infect Dis. 1992; 166: 35-41.
- Kleemann P, Distler E, Wagner EM, Thomas S, Klobuch S, et al. Varicellazoster virus glycoproteins B and E are major targets of CD4+ and CD8+ T cells reconstituting during zoster after allogeneic transplantation. Haematologica. 2012; 97: 874-82.
- Distler E, Schnürer E, Wagner E, von Auer C, Plachter B, et al. Recovery of varicella-zoster virus-specific T cell immunity after T cell-depleted allogeneic transplantation requires symptomatic virus reactivation. Biol Blood Marrow Transplant. 2008; 14: 1417-1424. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19041065
- Diaz PS, Smith S, Hunter E, Arvin AM. T lymphocyte cytotoxicity with natural varicella-zoster virus infection and after immunization with live attenuated varicella vaccine. J Immunol. 1989; 142: 636-641. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2536059
- Laing KJ, Russell RM, Dong L, Schmid DS, Stern M, et al. Zoster vaccination increases the breadth of CD4+ T cells responsive to varicella zoster virus. J Infect Dis. 2015; 212: 1022-1031. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25784732
- Haberthur K, Engelmann F, Park B, Barron A, Legasse A, et al. CD4 T cell immunity is critical for the control of simian varicella virus infection in a nonhuman primate model of VZV infection. PLoS Pathog. 2011; 7: e1002367. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22102814
- van der Heiden PL, de Boer R, van der Steen DM, Kester MG, van der Hoorn MW, et al. Identification of varicella-zoster virus-specific CD8 T cells in patients after T-cell-depleted allogeneic stem cell transplantation. J Virol. 2009; 83: 7361-7364. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19386715
- Arvin AM, Sharp M, Smith S, Koropchak CM, Diaz PS, et al. Equivalent recognition of a varicella-zoster virus immediate early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J Immunol. 1991; 146: 257-264. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1670603
- Jenkins DE, Yasukawa LL, Bergen R, Benike C, Engleman EG, et al. Comparison of primary sensitization of naive human T cells to varicella-zoster virus peptides by dendritic cells in vitro with responses elicited in vivo by varicella vaccination. J Immunol. 1999; 162: 560-567. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9886433
- Merindol N, Salem Fourati I, Brito RM, Grenier AJ, Charrier E, et al. Reconstitution of protective immune responses against cytomegalovirus and varicella zoster virus does not require disease development in pediatric recipients of umbilical cord blood transplantation. J Immunol. 2012; 189: 5016-5028. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23034171
- Kennedy JJ, Steain M, Slobedman B, Abendroth A. Infection and functional modulation of human monocytes and macrophages by varicella-zoster virus. J Virol. 2019; 93: e01887-18. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30404793
- Jones D, Como CN, Jing L, Blackmon A, Neff CP, et al. Varicella zoster virus productively infects human peripheral blood mononuclear cells to modulate expression of immunoinhibitory proteins and blocking PD-L1 enhances virusspecific CD8+ T cell effector function. PLoS Pathog. 2019; 15: e1007650. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30870532
- White TM, Gilden DH. Varicella virus-mononuclear cell interaction. Adv Virus Res. 2003; 62: 1-17. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3035815
- Gilden DH, Hayward AR, Krupp J, Hunter-Laszlo M, Huff JC, et al. Varicella-zoster virus infection of human mononuclear cells. Virus Res. 1987; 7: 117-129. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3035815
- Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, et al. Varicellazoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol. 2005; 79: 12658-12666. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16188968
- Yu HR, Huang HC, Kuo HC, Sheen JM, Ou CY, et al. IFN-α production by human mononuclear cells infected with varicella-zoster virus through TLR9dependent and -independent pathways. Cell Mol Immunol. 2011; 8: 181-188. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21317915
- Baba M, Shigeta S. Incomplete growth of varicella-zoster virus in human monocytes. Microbiol Immunol. 1983; 27: 767-777. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/6316116
- Devlin ME, Gilden DH, Mahalingam R, Dueland AN, Cohrs R. Peripheral blood mononuclear cells of the elderly contain varicella-zoster virus DNA. J Infect. Dis. 1992; 165: 619-622. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1313066
- Ordoñez G, Pineda B, Garcia-Navarrete R, Sotelo J. Brief presence of varicella-zoster viral DNA in mononuclear cells during relapses of multiple sclerosis. Arch Neurol. 2004; 61: 529-532. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15096401
- Vafai A, Wellish M, Gilden DH. Expression of varicella-zoster virus in blood mononuclear cells of patients with postherpetic neuralgia. Proc Natl Acad Sci USA. 1988; 85(8): 2767-2770. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2833752
- Gilden DH, Devlin M, Wellish M, Mahalingham R, Huff C, et al. Persistence of varicella-zoster virus DNA in blood mononuclear cells of patients with varicella or zoster. Virus Genes. 1989; 2: 299-305. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2554580
- Visalli MA, House BL, Selariu A, Zhu H, Visalli RJ. The varicella-zoster virus portal protein is essential for cleavage and packaging of viral DNA. J Virol. 2014; 88: 7973-7986. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24807720
- Riva L, Thiry M, Bontems S, Joris A, Piette J, et al. ORF9p phosphorylation by ORF47p is crucial for the formation and egress of varicella-zoster virus viral particles. J Virol. 2013; 87: 2868-2881. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23269791
- González-Motos V, Jürgens C, Ritter B, Kropp KA, Durán V, et al. Varicella zoster virus glycoprotein C increases chemokine-mediated leukocyte migration. PLoS Pathog. 2017; 13: e1006346. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28542541
- Xia D, Srinivas S, Sato H, Pesnica L, Straus SE, et al. Varicella-zoster virus open reading frame 21, which is expressed during latency, is essential for virus replication but dispensable for establishment of latency. J Virol. 2003; 77: 1211–1218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12502838
- Gary L, Gilden DH, Cohrs RJ. Epigenetic regulation of varicella-zoster virus open reading frames 62 and 63 in latently infected human trigeminal ganglia. J Virol. 2006; 80: 4921-4926. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16641283
- Cohen JI, Cox E, Pesnicak L, Srinivas S, Krogmann T. The varicella-zoster virus open reading frame 63 latency-associated protein is critical for establishment of latency. J Virol. 2004; 78: 11833-11840. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15479825
- Gerada C, Steain M, McSharry BP, Slobedman B, Abendroth A. Varicellazoster virus ORF63 protects human neuronal and keratinocyte cell lines from apoptosis and changes its localization upon apoptosis induction. J Virol. 2018; 92: e00338-18. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29593042
- Walters MS, Kyratsous CA, Silverstein SJ. The RING finger domain of varicella-zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J Virol. 2010; 84: 6861-6865. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20392849
- Epub 2010 Apr 14. 339. Zhang Z, Selariu A, Warden C, Huang G, Huang Y, et al. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 2010; 6 (7): e1000971. DOI: 10.1371/journal.ppat.1000971.
- Cohrs RJ, Lee KS, Beach A, Sanford B, Baird NL, et al. Targeted genome sequencing reveals varicella-zoster virus open reading frame 12 deletion. J Virol. 2017; 91: e01141-17. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28747504
- Vizoso Pinto MG, Pothineni VR, Haase R, Woidy M, Lotz-Havla AS, et al. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J Proteome Res. 2011; 10: 5374-5382. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21988664
- Lebrun M, Lambert J, Riva L, Thelen N, Rambout X, et al. Varicella-zoster virus ORF9p binding to cellular adaptor protein Complex 1 is important for viral infectivity. J Virol. 2018; 92: e00295-18. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29793951
- Oliver SL, Yang E, Arvin AM. Varicella-zoster virus glycoproteins: entry, replication, and pathogenesis. Curr Clin Microbiol Rep. 2016; 3: 204-215. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28367398
- Yang E, Arvin AM, Oliver SL. The glycoprotein B cytoplasmic domain lysine cluster is critical for varicella-zoster virus cell-cell fusion regulation and infection. J Virol. 2016; 91: e01707-1716. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27795427
- Yamagishi Y, Sadaoka T, Yoshii H, Somboonthum P, Imazawa T, et al. Varicella-zoster virus glycoprotein M homolog is glycosylated, is expressed on the viral envelope, and functions in virus cell-to-cell spread. J Virol. 2008; 82: 795-804. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17977964
- Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, et al. A site of varicellazoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci USA. 2015; 112: 6056-6061. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25918416
- Tavalai N, Stamminger T. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses. 2009; 1: 1240-1264. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21994592
- Hadjimichael C, Chanoumidou K, Nikolaou C, Klonizakis A, Theodosi GI, et al. Promyelocytic leukemia protein is an essential regulator of stem cell pluripotency and somatic cell reprogramming. Stem Cell Reports. 2017; 8: 1366-1378. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28392218
- Nakahara F, Weiss CN, Ito K. The role of PML in hematopoietic and leukemic stem cell maintenance. Int J Hematol. 2014; 100: 18-26. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24488785
- Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, et al. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 2011; 7: e1002157. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21901090
- Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010; 2: a000661. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20452955
- Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, et al. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog. 2011; 7: e1001266. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21304940
- El Asmi F, Maroui MA, Dutrieux J, Blondel D, Nisole S, et al. Implication of PMLIV in both intrinsic and innate immunity. PLoS Pathog. 2014; 10: e1003975. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24586174
- Maarifi G, Chelbi-Alix MK, Nisole S. PML control of cytokine signaling. Cytokine Growth Factor Rev. 2014; 25: 551-561. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24861946
- Hristozova N, Tompa P, Kovacs D. A novel method for assessing the chaperone activity of proteins. PLoS One. 2016; 11: e0161970. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27564234
- Livingston CM, Ifrim MF, Cowan AE, Weller SK. Virus-induced chaperoneenriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 2009; 5: e1000619. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/19816571
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013; 82: 323-355. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/23746257
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperonemediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004; 5: 781-791. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/15459659
- Semrad K. Proteins with RNA chaperone activity: a world of diverse proteins with a common task-impediment of RNA misfolding. Biochem Res Int. 2011; 2011: 532908. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/21234377
- Horwich AL, Weber-Ban EU, Finley D. Chaperone rings in protein folding and degradation. Proc Natl Acad Sci USA. 1999; 96: 11033-11040. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/10500119
- Kyratsous CA, Silverstein SJ. BAG3, a host cochaperone, facilitates varicellazoster virus replication. J Virol. 2007; 81: 7491-7503. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/17475647
- Knipe DM, Raja P, Lee JS. Clues to mechanisms of herpesviral latent infection and potential cures. Proc Natl Acad Sci USA. 2015; 112: 11993-11994. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26392531
- Li Q, Buranathai C, Grose C, Hutt-Fletcher LM. Chaperone functions common to nonhomologous Epstein-Barr virus gL and varicella-zoster virus gL proteins. J Virol. 1997; 71: 1667-1670. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/8995697
- Attar N. Viral infection: de-chaperoning antivirals. Nat Rev Microbiol. 2016; 14: 2. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26656092
- Eizuru Y. Development of new antivirals for herpesviruses. Antivir Chem Chemother. 2003; 14: 299-308. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/14968936
- Varadaraj A, Mattoscio D, Chiocca S. SUMO Ubc9 enzyme as a viral target. IUBMB Life. 2014; 66: 27-33. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24395713
- Lowrey AJ, Cramblet W, Bentz GL. Viral manipulation of the cellular sumoylation machinery. Cell Commun Signal. 2017; 15: 27. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28705221
- Mattoscio D, Segré CV, Chiocca S. Viral manipulation of cellular protein conjugation pathways: The SUMO lesson. World J Virol. 2013; 2: 79-90. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24175232
- Wimmer P, Schreiner S. Viral mimicry to usurp ubiquitin and SUMO host pathways. Viruses. 2015; 7: 4854-4872. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26343706
- Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010; 428: 133-145. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/20462400
- Sriramachandran AM, Dohmen RJ. SUMO-targeted ubiquitin ligases. Biochim Biophys Acta. 2014; 1843: 75-85. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24018209
- Sohn SY, Hearing P. Adenovirus early proteins and host sumoylation. MBio. 2016; 7: e01154-16. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27651358
- Kumar A, Zhang KY. Advances in the development of SUMO specific protease (SENP) inhibitors. Comput Struct Biotechnol J. 2015; 13: 204-211. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/25893082
- Stallings CL, Silverstein SJ. Posttranslational modification and cell typespecific degradation of varicella-zoster virus ORF29p. J Virol. 2006; 80: 10836-10846. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/16956951
- Rajsbaum R, García-Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. 2014; 426: 1265-1284. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24333484
- Adorisio S, Fierabracci A, Muscari I, Liberati AM, Ayroldi E, et al. SUMO proteins: Guardians of immune system. J Autoimmun. 2017; 84: 21-28. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28919255
- Grundhoff A, Sullivan CS. Virus-encoded microRNAs. Virology. 2011; 411: 325-343. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/21277611
- Li X, Huang Y, Zhang Y, He N. Evaluation of microRNA expression in patients with herpes zoster. Viruses. 2016; 8. E326. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27918431
- Markus A, Golani L, Ojha NK, Borodiansky-Shteinberg T, Kinchington PR, et al. Varicella-zoster virus expresses multiple small noncoding RNAs. J Virol. 2017; 91. e01710-e01717. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/29021397
- Kincaid RP, Sullivan CS. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog. 2012; 8: e1003018. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/23308061
- Qi Y, Zhu Z, Shi Z, Ge Y, Zhao K, et al. Dysregulated microRNA expression in serum of non-vaccinated children with varicella. Viruses. 2014; 6: 1823-1836. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24759212
- Jones M, Dry IR, Frampton D, Singh M, Kanda RK, et al. RNA-seq analysis of host and viral gene expression highlights interaction between varicella zoster virus and keratinocyte differentiation. PLoS Pathog. 2014; 10: e1003896. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24497829
- Henderson HH, Timberlake KB, Austin ZA, Badani H, Sanford B, et al. Occupancy of RNA polymerase II phosphorylated on serine 5 (RNAP S5P) and RNAP S2P on varicella-zoster virus genes 9, 51, and 66 is independent of transcript abundance and polymerase location within the gene. J Virol. 2015; 90: 1231-1243. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26559844
- Koniusz S, Andrzejewska A, Muraca M, Srivastava AK, Janowski M, et al. Extracellular vesicles in physiology, pathology, and therapy of the immune and central nervous system, with focus on extracellular vesicles derived from mesenchymal stem cells as therapeutic tools. Front Cell Neurosci. 2016; 10: 109. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27199663
- Bello-Morales R, López-Guerrero JA. Extracellular vesicles in herpes viral spread and immune evasion. Front Microbiol. 2018; 9: 2572. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30410480
- Pleet ML, Branscome H, DeMarino C, Pinto DO, Zadeh MA, et al. Autophagy, EVs, and infections: A perfect question for a perfect time. Front Cell Infect Microbiol. 2018; 8: 362. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30406039
- Liu L, Zhou Q, Xie Y, Zuo L, Zhu F, et al. Extracellular vesicles: novel vehicles in herpesvirus infection. Virol Sin. 2017; 32: 349-356. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/29116589
- Crenshaw BJ, Gu L, Sims B, Matthews QL. Exosome biogenesis and biological function in response to viral infections. Open Virol J. 2018; 12: 134-148. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30416610
- Anderson MR, Kashanchi F, Jacobson S. Exosomes in viral disease. Neurotherapeutics. 2016; 13: 535-546. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27324390
- Sampey GC, Meyering SS, Zadeh MA, Saifuddin M, Hakami RM, et al. Exosomes and their role in CNS viral infections. J Neurovirol. 2014; 20: 199-208. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/24578033
- Alenquer M, Amorim MJ. Exosome biogenesis, regulation, and function in viral infection. Viruses. 2015; 7: 5066-5083. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26393640
- Ferreira VL, Borba H, Bonetti AF, Leonart LP, Pontarolo R. Cytokines and interferons: types and functions. In: Autoantibodies and cytokines.
- Zajkowska A, Garkowski A, Świerzbińska R, Kułakowska A, Król ME, et al. Evaluation of chosen cytokine levels among patients with herpes zoster as ability to provide immune response. PLoS One. 2016; 11: e0150301. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26934574
- Cornaby C, Tanner A, Stutz EW, Poole BD, Berges BK. Piracy on the molecular level: human herpesviruses manipulate cellular chemotaxis. J Gen Virol. 2016; 97: 543-560. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26669819
- Pontejo SM, Murphy PM, Pease JE. Chemokine subversion by human herpesviruses. J Innate Immun. 2018; 10: 465-478. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30165356
- Torres-Pedraza S, Betancur JG, Urcuqui-Inchima S. Viral recognition by the innate immune system: the role of pattern recognition receptors. Colomb Méd. 2010; 41: 377-387.
- Jenkins DE, Redman RL, Lam EM, Liu C, Lin I, et al. Interleukin (IL)-10, IL12, and interferon-gamma production in primary and memory immune responses to varicella-zoster virus. J Infect Dis. 1998; 178: 940-948. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/9806019
- Zak-Prelich M, McKenzie RC, Sysa-Jedrzejowska A, Norval M. Local immune responses and systemic cytokine responses in zoster: relationship to the development of postherpetic neuralgia. Clin Exp Immunol. 2003; 131: 318-323. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/12562395
- Jarosinski KW, Carpenter JE, Buckingham EM, Jackson W, Knudtson K, et al. Cellular stress response to varicella-zoster virus infection of human skin includes highly elevated interleukin-6 xpression. Open Forum Infect Dis. 2018; 5: ofy118. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30014002
- Jones D, Neff CP, Palmer BE, Stenmark K, Nagel MA. Varicella zoster virusinfected cerebrovascular cells produce a proinflammatory environment. Neurol Neuroimmunol Neuroinflamm. 2017; 4: e382. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/29159203
- Choi EJ, Lee CH, Shin OS. Suppressor of cytokine signaling 3 expression induced by varicella-zoster virus infection results in the modulation of virus replication. Scand J Immunol. 2015; 82: 337-344. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26072679
- Arvin AM, Koropchak CM, Williams BR, Grumet FC, Foung SK. Early immune response in healthy and immunocompromised subjects with primary varicella-zoster virus infection. J Infect Dis. 1986; 154: 422-429. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/3016110
- Smith-Norowitz TA, Josekutty J, Lev-Tov H, Kohlhoff S, Norowitz KB, et al. IgE anti-varicella zoster virus and other immune responses before, during, and after shingles. Ann Clin Lab Sci. 2009; 39: 43-50. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/19201740
- Nour AM, Reichelt M, Ku CC, Ho MY, Heineman TC, et al. Varicella-zoster virus infection triggers formation of an interleukin-1β (IL-1β)-processing inflammasome complex. J Biol Chem. 2011; 286: 17921-1733. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/21385879
- Fujimura T, Yamanashi R, Masuzawa M, Fujita Y, Katsuoka K, et al. Conversion of the CD4+ T cell profile from T(H2)-dominant type to T(H1)dominant type after varicella-zoster virus infection in atopic dermatitis. J Allergy Clin Immunol. 1997; 100: 274-282. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/9275152
- Bayat A, Burbelo PD, Browne SK, Quinlivan M, Martinez B, et al. Anti-cytokine autoantibodies in postherpetic neuralgia. J Transl Med. 2015; 13: 333. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26482341
- Zhu SM, Liu YM, An ED, Chen QL. Influence of systemic immune and cytokine responses during the acute phase of zoster on the development of postherpetic neuralgia. J Zhejiang Univ Sci B. 2009; 10: 625-630. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/19650202
- Hao M, Wang X, Du J, Liu L, Jiao Y, et al. Cytokine levels are associated with the severity of varicella infections. J Infect Dev Ctries. 2015; 9: 190-196. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/25699494
- Bubak AN, Como CN, Blackmon AM, Jones D, Nagel MA. Varicella zoster virus differentially alters morphology and suppresses proinflammatory cytokines in primary human spinal cord and hippocampal astrocytes. J Neuroinflammation. 2018; 15: 318. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30442152
- Lind L, Eriksson K, Grahn A. Chemokines and matrix metalloproteinases in cerebrospinal fluid of patients with central nervous system complications caused by varicella-zoster virus. J Neuroinflammation. 2019; 16: 42. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/30777092
- de Visser L, H de Boer J, T Rijkers G, Wiertz K, van den Ham HJ, et al. Cytokines and chemokines involved in acute retinal necrosis. Invest Ophthalmol Vis Sci. 2017; 58: 2139-2151. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28395298
- Graybill C, Claypool DJ, Brinton JT, Levin MJ, Lee KS. Cytokines produced in response to varicella-zoster virus infection of ARPE-19 cells stimulate lymphocyte chemotaxis. J Infect Dis. 2017; 216: 1038-1047. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28968855
- Traina-Dorge V, Sanford R, James S, Doyle-Meyers LA, de Haro E, et al. Robust pro-inflammatory and lesser anti-inflammatory immune responses during primary simian varicella virus infection and reactivation in rhesus macaques. J Neurovirol. 2014; 20: 526-530. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/25139181
- Otani N, Baba K, Okuno T. Interferon-gamma release assay: a simple method for detection of varicella-zoster virus-specific cell-mediated immunity. J Immunol Methods. 2009; 351: 71-74. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/19818791
- Verweij MC, Wellish M, Whitmer T, Malouli D, Lapel M, et al. Varicella viruses inhibit interferon-stimulated JAK-STAT signaling through multiple mechanisms. PLoS Pathog. 2015; 11: e1004901. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/25973608
- Rahaus M, Desloges N, Wolff MH. Varicella-zoster virus influences the activities of components and targets of the ERK signalling pathway. J Gen Virol. 2006; 87: 749-758. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/16528022
- Kurapati S, Sadaoka T, Rajbhandari L, Jagdish B, Shukla P, et al. Role of the JNK pathway in varicella-zoster virus lytic infection and eactivation. J Virol. 2017; 91: e00640-17. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28637759
- Rahaus M, Desloges N, Wolff MH. Varicella-zoster virus requires a functional PI3K/Akt/GSK-3alpha/beta signaling cascade for efficient replication. Cell Signal. 2007; 19: 312-320. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/16934436
- François S, Sen N, Mitton B, Xiao X, Sakamoto KM, et al. Varicella-zoster virus activates CREB, and inhibition of the pCREB-p300/CBP interaction inhibits viral replication in vitro and skin pathogenesis in vivo. J Virol. 2016; 90: 86868697. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27440893
- Neirinckx V, Coste C, Rogister B, Wislet-Gendebien S. Neural fate of mesenchymal stem cells and neural crest stem cells: which ways to get neurons for cell therapy purpose? 2013.
- Chi PI, Liu HJ. Molecular signaling and cellular pathways for virus entry. ISRN Virol. 2013; 8: 306595.
- Rahaus M, Desloges N, Wolff MH. ORF61 protein of varicella-zoster virus influences JNK/SAPK and p38/MAPK phosphorylation. J Med Virol. 2005; 76: 424-433. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/15902710
- Fleming SB. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines (Basel). 2016; 4. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/27367734
- Kuchipudi SV. The complex role of STAT3 in viral infections. J Immunol Res. 2015; 2015: 272359. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26199948
- Roca Suarez AA, Van Renne N, Baumert TF, Lupberger J. Viral manipulation of STAT3: evade, exploit, and injure. PLoS Pathog. 2018; 14: e1006839. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/29543893
- Sen N, Che X, Rajamani J, Zerboni L, Sung P, et al. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci USA. 2012; 109: 600-605. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/22190485
- Ku CC, Chang YH, Chien Y, Lee TL. Type I interferon inhibits varicellazoster virus replication by interfering with the dynamic interaction between mediator and IE62 within replication compartments. Cell Biosci. 2016; 6: 21. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/26985360
- Ouwendijk WJD, van Veen S, Mahalingam R, Verjans GMGM. Simian varicella virus inhibits the interferon gamma signalling pathway. J Gen Virol. 2017; 98: 2582-2588. PubMed.: https://www.ncbi.nlm.nih.gov/pubmed/28901902
- Shakya AK, O'Callaghan DJ, Kim SK. Interferon gamma inhibits varicellazoster virus replication in a cell line-dependent manner. J Virol. 2019; 93. 00257-00319.
- Zwezdaryk KJ, Combs JA, Morris CA, Sullivan DE. Regulation of Wnt/βcatenin signaling by herpesviruses. World J Virol. 2016; 5: 144-154.