
More Information
Submitted: March 27, 2023 | Approved: April 14, 2023 | Published: April 17, 2023
How to cite this article: Patel UV, Sangha N, Rettew A. Systemic sclerosis sine scleroderma presenting as renal crisis, a case report and review of the literature. J Hematol Clin Res. 2023; 7: 006-010.
DOI: 10.29328/journal.jhcr.1001021
Copyright License: © 2023 Patel UV, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Systemic sclerosis sine scleroderma; Scleroderma renal crisis; Thrombotic microangiopathy

Systemic sclerosis sine scleroderma presenting as renal crisis, a case report and review of the literature
Urvi V Patel, Navdeep Sangha* and Andrew Rettew
Department of Internal Medicine, Tower Health Reading Hospital, 420 South Fifth Avenue, West Reading, PA 19611, USA
*Address for Correspondence: Navdeep Sangha, DO, Department of Internal Medicine, Tower Health Reading Hospital, 420 South Fifth Avenue, West Reading, PA 19611, USA, Email: [email protected]
Systemic sclerosis sine scleroderma is a rare subset of systemic sclerosis with isolated organ involvement. Scleroderma renal crisis is a severe manifestation of systemic sclerosis characterized by malignant hypertension, oligo/anuric renal failure, and thrombotic microangiopathy. We present a case of a 55-year-old male with uncontrolled hypertension who presented with hematospermia and was found to have acute renal failure, microangiopathic hemolytic anemia, concerning thrombotic microangiopathy. Empiric management for thrombotic thrombocytopenic purpura (TTP) with plasma exchange and corticosteroids yielded a paradoxical response, ultimately leading to the diagnosis of systemic sclerosis sine scleroderma presenting as scleroderma renal crisis (SRC) after serological confirmation. Given the morbidity and mortality associated with scleroderma renal crisis, it should be increasingly considered as a differential for thrombotic microangiopathy even without outward manifestations of systemic sclerosis. Additionally, the empiric management of TTP can include the use of corticosteroids which can exacerbate SRC, an early clinical clue in the diagnosis of this disease.
Systemic sclerosis is a rare multi-organ autoimmune disease with a prevalence of 275 cases per million [1]. It can be classified into limited systemic sclerosis, diffuse systemic sclerosis, systemic sclerosis sine scleroderma (ssSSc), and overlap forms of the disease. The 2013 European League against Rheumatism (EULAR) criteria for systemic sclerosis are used to classify patients as having limited or diffuse cutaneous SSc.
These criteria are:
1. Skin thickening of the fingers extending proximal to the metacarpophalangeal joints
2. Skin thickening of the hands extending proximal to the wrist.
3. Skin thickening of the face.
4. Interstitial lung disease or pulmonary arterial hypertension.
5. Raynaud’s phenomenon.
6. Sclerodactyly (thickening and tightening of the skin on the fingers).
7. Telangiectasia (dilated blood vessels) on the hands and face.
A patient is considered to have diffuse cutaneous SSc if they have skin thickening of the trunk, skin thickening of the proximal extremities (excluding the fingers), or skin thickening of the face. A patient is considered to have limited cutaneous SSc if they do not meet the criteria for diffuse cutaneous SSc but have either Raynaud’s phenomenon or telangiectasia.
Systemic sclerosis sine scleroderma is a rare subset of scleroderma seen in only 2-9% of patients and is defined by isolated organ involvement in the absence of skin fibrosis [2,3]. The EULAR does not have a specific definition for SScSS, but it is generally diagnosed based on the absence of skin thickening or tightening that is characteristic of SSc. However, patients with SScSS may have other signs of the disease, such as pulmonary arterial hypertension, gastrointestinal or renal involvement, and autoantibody positivity, which can aid in the diagnosis, as in our patient.
Scleroderma renal crisis (SRC) occurs in 5% - 13% of all systemic sclerosis cases and in 12% - 33% of patients with RNA polymerase III positivity [4]. Although there is no universally accepted definition of SRC, it is characterized by non-oliguric rapid renal failure and acute onset hypertension with a marked increase in systemic blood pressure (> 30 mmHg systolic and > 20 mmHg diastolic above baseline) [4]. Approximately 40% - 50% of SRC cases present with microangiopathy [5]. The pathogenesis of SRC is unclear with some studies suggesting vasculopathic changes in the kidneys resulting in the thickening of vessel walls while other studies suggest complement pathway dysregulation [5,6]. Corticosteroids are a well-recognized trigger of scleroderma renal crisis [5]. The associated morbidity and mortality in SRC are high with 40% - 50% of patients requiring hemodialysis and a 5-year mortality rate of 50% - 70% [4]. This case report highlights the importance of keeping a broad differential when clinicians are presented with thrombotic microangiopathies and the challenges associated with the empiric management of this rare but often fatal disease.
We present to you a case of a 55-year-old male with hypertension and no history of Raynaud’s phenomenon who presented to the hospital with gastroesophageal reflux symptoms and hematospermia with dark urine. On initial evaluation, the patient was afebrile and tachycardic to 100 beats per minute with a blood pressure of 173/97 mmHg while saturating on ambient air. Physical examination was notable for scleral icterus but otherwise unremarkable. Initial blood chemistries showed a creatinine of 8.39 mg/dL compared to a prior baseline of 0.89 mg/dL and blood urea nitrogen of 84 mg/dL. Hemogram was remarkable for hemoglobin of 9.4 g/dL and platelets of 82,000/mm3. Hemolysis labs showed a significantly elevated lactate dehydrogenase at 752 IU/L (normal 140-271 IU/L), and a reticulocyte count of 4% (normal 0.5% to 1.5%). A direct Coombs test was negative. Haptoglobin was less than 30 mg/dL (normal 44 - 215 mg/dL), fibrinogen was 423 mg/dL (normal 222 - 525 mg/dL) and d-dimer was 0.80 mcg/mL. A peripheral blood smear demonstrated decreased platelet count without clumping and numerous schistocytes. Urinalysis showed moderate blood with 30 mg/dL of protein. Urine toxicology was negative. Initial imaging studies included chest radiographs and computed tomography of the chest and showed no acute abnormalities. A renal ultrasound showed a left renal cyst but no evidence of hydronephrosis. An echocardiogram showed normal left ventricular systolic function, Grade 1 diastolic dysfunction, and a small-size pericardial effusion.
Given concern for thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS), the patient was empirically started on plasma exchange and received three doses of intravenous methylprednisolone followed by a prednisone taper. Stool studies were negative for Escherichia Coli O157, Enterohemorrhagic Escherichia coli, Salmonella, and Shigella. Stool studies incidentally came back positive for Campylobacter. An ADAMTS13 level was ordered and later resulted in a level of 0.6 IU/mL (normal 0.68 - 1.63 IU/mL).
The patient’s hospital course was complicated by flash pulmonary edema and hypertensive emergency requiring nicardipine drip and subsequent antihypertensive therapy with hydralazine, carvedilol, and nifedipine. The patient had improvement in hemolytic parameters after treatment with blood pressure control, corticosteroids, and plasma exchange but had no improvement in renal function, ultimately requiring hemodialysis. He subsequently underwent a renal biopsy which revealed thrombotic microangiopathy with a spectrum of acute and subacute lesions including overt fibrin thrombosis, ischemic glomerular changes, and severe vascular narrowing with intimal edema, fibrin, and schistocytes (Figures 1-3).
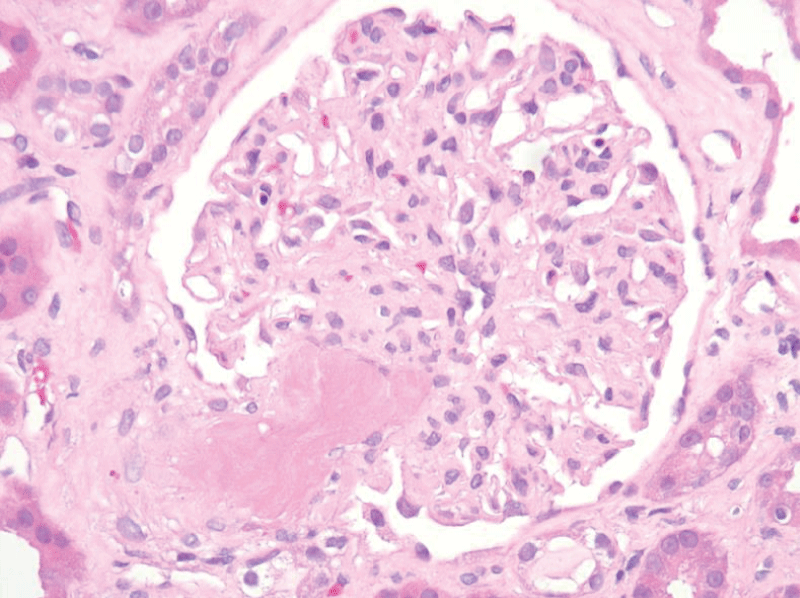
Figure 1: A surgical biopsy of the left kidney showed a fibrin thrombus at the vascular pole.
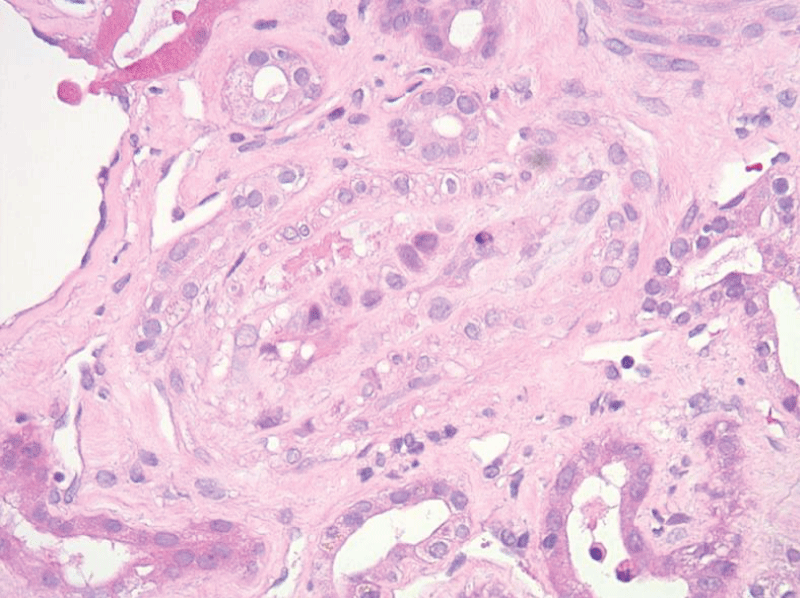
Figure 2: A surgical biopsy of the left kidney showed a small artery with intimal swelling and fibrin.
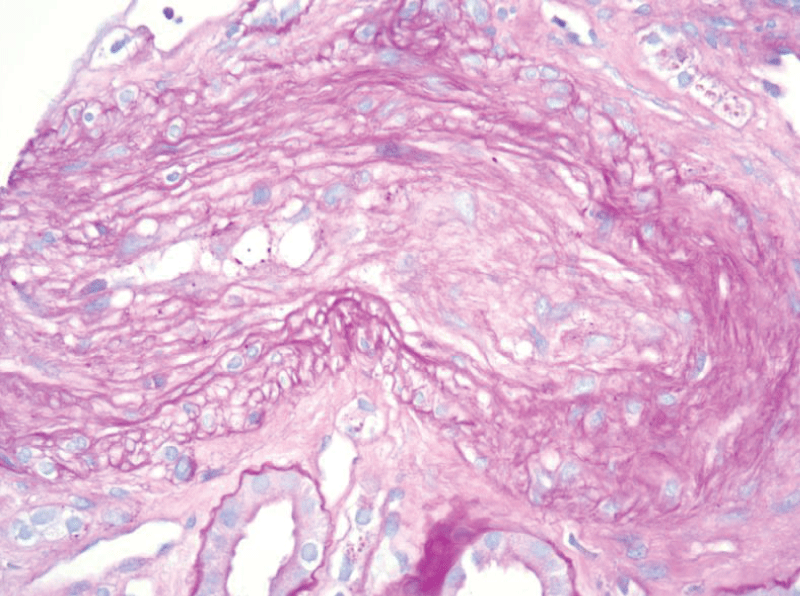
Figure 3: A surgical biopsy of the left kidney showed arterial onion skinning.
At this juncture, the patient was considered for eculizumab therapy for a suspected diagnosis of complement-mediated thrombotic microangiopathy given negative stool studies and an ADAMTS13 level above 10%. As such the patient received prophylactic meningococcal vaccinations prior to the administration of eculizumab. The patient received one dose of eculizumab inpatient and was discharged with the aforementioned antihypertensives, prednisone taper, and outpatient follow-up with hematology, nephrology, and rheumatology.
He ultimately received only one dose of eculizumab as the atypical HUS gene panel was obtained but later found to be equivocal. Anti-complement factor H autoantibodies were positive at 41 U (normal < 20 U). Additional rheumatologic workup was obtained and pending results prior to discharge given renal biopsy findings. Rheumatologic studies (Table 1) revealed an antinuclear antibody titer of 1:320 (normal < 1:40) with a nuclear speckled pattern, Anti-Ro antibody > 8 AI (normal < 1 AI), and RNA polymerase III antibody positive at 63 U (normal < 20 U) (Table 1). Negative rheumatologic investigations included anti-centromere antibodies, anti-neutrophil cytoplasmic antibodies, complement C3, complement C4, myeloperoxidase antibody, anti-LA antibody (SS-B), anticardiolipin IgM and IgG, antinuclear ribonucleoprotein antibody, anti-double-stranded DNA antibody, B2 glycoprotein, lupus anticoagulant, anti-smith antibody, anti-topoisomerase-1 antibody (SCL-70), anti-glomerular basement membrane antibody, and complement C1Q. During the hospitalization, the patient was also found to have a vitamin B12 deficiency, and intramuscular supplementation was given. Later pernicious anemia was diagnosed given a positive intrinsic factor-blocking antibody.
| Table 1: Rheumatologic Investigations. | ||
| Study | Result | Reference Value |
| Antinuclear antibody | 1: 320. Nuclear Speckled Pattern | < 1: 40 |
| ANCA | Negative | Negative |
| C3 | 88 mg/dL | 82 - 185 mg/dL |
| C4 | 32 mg/dL | 15 - 53 mg/dL |
| Complement C1q | 7.1 mg/dL | 5.0 - 8.6 mg/dL |
| Anti - Complement Factor H Antibody (CFH) | 41 U | < 20 U |
| Myeloperoxidase antibody (MPO) | < 1.0 AI | < 1.0 AI |
| Anti-glomerular basement membrane antibody (GBM) | < 1.0 AI | < 1.0 AI |
| Anti-double-stranded DNA antibody | < 1.0 IU/mL | < 4.0 IU/mL |
| Autoantibody to Smith | < 1.0 AI | < 1.0 AI |
| Anti-Ro Antibody (SS-A) | > 8 AI | < 1 AI |
| Anti - La Antibody (SS-B) | < 1.0 AI | < 1.0 AI |
| Centromere B Antibody | < 1.0 AI | < 1.0 AI |
| Anti-topoisomerase I antibody (SCL-70) | < 1.0 AI | < 1.0 AI |
| Anti - ribonucleoprotein antibody (RNP) | < 1.0 AI | < 1.0 AI |
| Anti-RNA Polymerase III ab | 63 U | < 20 U |
| Lupus anticoagulant | Not detected | Not detected |
| Beta-2 Glycoprotein IgG, IgM, and IgA | < 2.0 U/mL | < 20.0 U/mL |
| Anticardiolipin IgM and IgG | < 2.0 GPL-U/mL | < 20.0 GPL-U/mL |
| Parietal Cell antibody | < 20 U | < 20 U |
| Intrinsic Factor blocking Ab | Positive | Negative |
The patient was seen in follow-up in our hematology office 5 days after discharge. The positive RNA polymerase III antibody was consistent with a diagnosis of systemic sclerosis, however, given the absence of outward manifestations of scleroderma the patient was diagnosed with systemic sclerosis sine scleroderma presenting as scleroderma renal crisis. At this time his anti-hypertensive therapy was changed to include ACE inhibitors and prednisone was discontinued. A CT chest, abdomen, and pelvis were ordered to rule out any underlying malignancy and the presence of RNA polymerase III as a paraneoplastic phenomenon. He continued to undergo hemodialysis three times per week and was referred to rheumatology for systemic sclerosis management. He was referred to gastroenterology for esophagogastroduodenoscopy (EGD) for cancer screening and evaluation of SSc involvement of the gastric mucosa in the setting of pernicious anemia and advised to continue parenteral vitamin B12 supplementation.
Here we presented a diagnostically challenging case of scleroderma renal crisis sine scleroderma that presented with gastrointestinal symptoms, uncontrolled hypertension, acute renal failure requiring hemodialysis, and microangiopathic hemolytic anemia. These features were a red flag for thrombotic microangiopathy, initially thought to be TTP or complement-mediated thrombotic microangiopathy (CM-TMA). Scleroderma renal crisis was not considered as part of the initial differential diagnosis as the patient did not have any features of systemic sclerosis or prior history of Raynaud’s phenomenon. Given the lack of improvement with plasmapheresis and glucocorticoids, CM-TMA then became a suspected diagnosis but corticosteroid-triggered scleroderma renal crisis was not considered until renal biopsy and RNA Polymerase III positivity.
This case highlights several important points. First, scleroderma renal crisis should be on the differential diagnosis for thrombotic microangiopathy despite the lack of cutaneous manifestations of systemic sclerosis as renal involvement can occur in 22% of ssSSc [7]. Second, this case demonstrates the importance of renal biopsy in distinguishing between the subtypes of thrombotic microangiopathy, as seen in our case. Kidney biopsy was crucial in guiding differential diagnosis as the pathologist noted that the overt fibrin thrombosis seen in our patient’s biopsy is not a frequent finding in TMA due to accelerated hypertension alone. Additionally, the prominent arterial findings in our patient’s kidney biopsy are not characteristic of what is typically seen in TTP. Thrombotic microangiopathy with a predilection for arteries and small vessels rather than glomeruli favors SRC over HUS or TTP [8,9]. While aHUS remained on the differential, the biopsy prompted further rheumatologic investigations including anti-topoisomerase I antibody, RNA polymerase III antibody, and anti-ribonucleoprotein antibody. The autoantibody positivity with isolated organ involvement of the kidney was essential to our diagnosis of ssSSc. An echocardiogram was performed as mentioned earlier but did not show any evidence of cardiac involvement such as pericarditis or myocardial fibrosis. A high-resolution computed tomography (CT) of the chest was never completed and would have aided in identifying any pleuritis, interstitial lung disease (ILD), or pulmonary veno-occlusive disease. ILD occurs in 50% of patients with DCSSc. Additionally, nail fold capillaroscopy should be considered in the future to evaluate for vascular involvement.
Third, although the pathogenesis of SRC is not well understood, this case supports existing theories that similarities between aHUS and SRC may exist with respect to complement-mediated injury. Recent literature suggests that complement activation plays an integral role in the development of SRC; further, there appears to be a growing body of evidence supporting complement activation-via both the alternative or classical pathway- as integral to the development of most thrombotic microangiopathies [10-12].
Factor H is one such key regulator of the alternative complement pathway. The impairment of factor H via the development of autoantibodies, for instance, has an established role in the development of TMAs. In fact, the presence of factor H autoantibodies has been noted in over 10% of aHUS cases [13,14]. Although genetic predisposition has been posited as one mechanism for developing these autoantibodies, acquired forms are also noted in the literature [14]. An atypical HUS genetic panel was ordered in this case and found to be equivocal. The patient had three polymorphisms in CFH which commonly appear together and are as prevalent as 23% in the healthy population but statistically enriched in the aHUS patient. The anti-CFH antibody was positive in this patient but was not high enough to be diagnostic for complement-mediated TMA. The presence of anti-CFH antibodies likely represents a consequence of the complement system being involved in the pathogenesis of the TMA observed in both disease states [6,15]. A gene screening study of patients with positive anti-RNA polymerase III who developed scleroderma renal crisis (SRC) also showed a strong association with the complement system [15]. Given this growing body of evidence supporting the role of complement in SRC, there have been case reports of using eculizumab, a terminal complement inhibitor, in its management; however, with only a limited degree of success [10,16].
Additionally, the concurrent diagnosis of pernicious anemia by virtue of auto-antibodies to intrinsic factor and parietal cells in an older adult with concurrent aforementioned antibodies is concerning for a diffuse autoimmune phenomenon driving the patient’s presentation. Metabolic TMA was thought to be unlikely in this patient as he did not have overt macrocytosis or improvement in the course with vitamin B12 supplementation. Cases of concurrent pernicious anemia with systemic sclerosis have also been reported in the literature, alongside SIBO-induced B12 deficiency, providing further support for this as the unifying diagnosis [17].
In summary, the patient’s course was most consistent with ssSSc with SRC given the low titer cross-reactive auto-antibodies, with a known association of complement activation and paradoxical response to corticosteroid therapy [18]. Once the patient was diagnosed with ssSSc with SRC, prednisone was discontinued, and the patient was maximized on ACE-inhibitor therapy. The patient is undergoing hemodialysis three times weekly however, he has not shown any significant renal recovery. As evidenced by this case, ssSSc with a renal crisis is diagnostically challenging and presents with significant morbidity to the patient requiring a broad differential at onset and a swift multidisciplinary approach. Further, we posit that perhaps this case with the presence of dual RNA Polymerase III and anti-CFH antibodies delivers insight into the mechanism by which complement activation is noted in SRC patients. If validated, this would provide meaningful guidance on the direction for future therapy beyond angiotensin inhibition for this highly fatal disease.
Ethical considerations
This case report was completed retrospectively after the patient had received appropriate treatment and care in the hospital. This case report did not require Institutional Review Board approval but was reviewed by the authors as follows. In order to protect the patient’s confidentiality, the authors reviewed the final manuscript and all personal identifiers were removed from the case report’s figures and special care was taken in the data contained in the description of the case so that the individual cannot be identified.
Data availability: Data sharing is not applicable to this article as no new data were created or analyzed in this study.
We thank Dr. Leal C. Herlitz from the Department of anatomic pathology at Cleveland Clinic for her contribution.
- McCray CJ, Mayes MD. Update on systemic sclerosis. Curr Allergy Asthma Rep. 2015 May;15(5):25. doi: 10.1007/s11882-015-0526-0. PMID: 26139334.
- Lee CM, Girnita D, Sharma A, Khanna S, Elwing JM. A Unique Presentation of Anti-RNA Polymerase III Positive Systemic Sclerosis Sine Scleroderma. Case Rep Rheumatol. 2016;2016:8536341. doi: 10.1155/2016/8536341. Epub 2016 Jul 31. PMID: 27559487; PMCID: PMC4983352.
- Sobolewski P, Maślińska M, Wieczorek M, Łagun Z, Malewska A, Roszkiewicz M, Nitskovich R, Szymańska E, Walecka I. Systemic sclerosis - multidisciplinary disease: clinical features and treatment. Reumatologia. 2019;57(4):221-233. doi: 10.5114/reum.2019.87619. Epub 2019 Aug 31. PMID: 31548749; PMCID: PMC6753596.
- Zanatta E, Polito P, Favaro M, Larosa M, Marson P, Cozzi F, Doria A. Therapy of scleroderma renal crisis: State of the art. Autoimmun Rev. 2018 Sep;17(9):882-889. doi: 10.1016/j.autrev.2018.03.012. Epub 2018 Jul 10. PMID: 30005860.
- Zanatta E, Codullo V, Allanore Y. Scleroderma renal crisis: Case reports and update on critical issues. Eur J Rheumatol. 2021 Jul;8(3):162-167. doi: 10.5152/eurjrheum.2020.20048. PMID: 33226326; PMCID: PMC9770404.
- Zuckerman R, Asif A, Costanzo EJ, Vachharajani T. Complement activation in atypical hemolytic uremic syndrome and scleroderma renal crisis: a critical analysis of pathophysiology. J Bras Nefrol. 2018 Jan-Mar;40(1):77-81. doi: 10.1590/2175-8239-JBN-3807. Epub 2018 May 7. PMID: 29796581; PMCID: PMC6533968.
- Hunzelmann N, Genth E, Krieg T, Lehmacher W, Melchers I, Meurer M, Moinzadeh P, Müller-Ladner U, Pfeiffer C, Riemekasten G, Schulze-Lohoff E, Sunderkoetter C, Weber M, Worm M, Klaus P, Rubbert A, Steinbrink K, Grundt B, Hein R, Scharffetter-Kochanek K, Hinrichs R, Walker K, Szeimies RM, Karrer S, Müller A, Seitz C, Schmidt E, Lehmann P, Foeldvári I, Reichenberger F, Gross WL, Kuhn A, Haust M, Reich K, Böhm M, Saar P, Fierlbeck G, Kötter I, Lorenz HM, Blank N, Gräfenstein K, Juche A, Aberer E, Bali G, Fiehn C, Stadler R, Bartels V; Registry of the German Network for Systemic Scleroderma. The registry of the German Network for Systemic Scleroderma: frequency of disease subsets and patterns of organ involvement. Rheumatology (Oxford). 2008 Aug;47(8):1185-92. doi: 10.1093/rheumatology/ken179. Epub 2008 May 31. PMID: 18515867; PMCID: PMC2468885.
- Gallan AJ, Chang A. A New Paradigm for Renal Thrombotic Microangiopathy. Semin Diagn Pathol. 2020 May;37(3):121-126. doi: 10.1053/j.semdp.2020.01.002. Epub 2020 Feb 6. PMID: 32085935.
- Tostivint I, Mougenot B, Flahault A, Vigneau C, Costa MA, Haymann JP, Sraer JD, Rondeau E. Adult haemolytic and uraemic syndrome: causes and prognostic factors in the last decade. Nephrol Dial Transplant. 2002 Jul;17(7):1228-34. doi: 10.1093/ndt/17.7.1228. PMID: 12105245.
- Devresse A, Aydin S, Le Quintrec M, Demoulin N, Stordeur P, Lambert C, Gastoldi S, Pirson Y, Jadoul M, Morelle J. Complement activation and effect of eculizumab in scleroderma renal crisis. Medicine (Baltimore). 2016 Jul;95(30):e4459. doi: 10.1097/MD.0000000000004459. PMID: 27472742; PMCID: PMC5265879.
- Chua JS, Baelde HJ, Zandbergen M, Wilhelmus S, van Es LA, de Fijter JW, Bruijn JA, Bajema IM, Cohen D. Complement Factor C4d Is a Common Denominator in Thrombotic Microangiopathy. J Am Soc Nephrol. 2015 Sep;26(9):2239-47. doi: 10.1681/ASN.2014050429. Epub 2015 Jan 8. PMID: 25573909; PMCID: PMC4552108.
- Teoh CW, Riedl M, Licht C. The alternative pathway of complement and the thrombotic microangiopathies. Transfus Apher Sci. 2016 Apr;54(2):220-31. doi: 10.1016/j.transci.2016.04.012. Epub 2016 Apr 26. PMID: 27160864.
- Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, Morgan L, Goodship TH, Marchbank KJ. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010 Jan 14;115(2):379-87. doi: 10.1182/blood-2009-05-221549. Epub 2009 Oct 27. PMID: 19861685; PMCID: PMC2829859.
- Zhang Y, Ghiringhelli Borsa N, Shao D, Dopler A, Jones MB, Meyer NC, Pitcher GR, Taylor AO, Nester CM, Schmidt CQ, Smith RJH. Factor H Autoantibodies and Complement-Mediated Diseases. Front Immunol. 2020 Dec 15;11:607211. doi: 10.3389/fimmu.2020.607211. PMID: 33384694; PMCID: PMC7770156.
- Guerra SG, Fonseca C, Nihtyanova SI, Stern E, Abraham DJ, Burns A, Harber M, Denton CP. Defining genetic risk for scleroderma renal crisis: a genome-wide analysis of anti-RNA polymerase antibody-positive systemic sclerosis. British Journal of Rheumatology. 2015; 54(1): 159-159. https://doi.org/10.1093/rheumatology/kev090.047.
- Okrój M, Johansson M, Saxne T, Blom AM, Hesselstrand R. Analysis of complement biomarkers in systemic sclerosis indicates a distinct pattern in scleroderma renal crisis. Arthritis Res Ther. 2016 Nov 18;18(1):267. doi: 10.1186/s13075-016-1168-x. PMID: 27863511; PMCID: PMC5116178.
- Hirose Y, Yoshida Y, Konda S, Tanaka N, Hirone T. Pernicious anemia in a patient with progressive systemic sclerosis. Nihon Ketsueki Gakkai Zasshi. 1977 Feb;40(1):1-8. PMID: 577644.
- Lee H, Kang E, Kang HG, Kim YH, Kim JS, Kim HJ, Moon KC, Ban TH, Oh SW, Jo SK, Cho H, Choi BS, Hong J, Cheong HI, Oh D. Consensus regarding diagnosis and management of atypical hemolytic uremic syndrome. Korean J Intern Med. 2020 Jan;35(1):25-40. doi: 10.3904/kjim.2019.388. Epub 2020 Jan 2. PMID: 31935318; PMCID: PMC6960041.