
More Information
Submitted: September 06, 2023 | Approved: September 19, 2023 | Published: September 20, 2023
How to cite this article: Kmira Z, Nedia S, Noura BY, Nesrine BS, Ahmed G, et al. A Rare Case of an Acquired Isolated Factor VII Deficiency was Discovered in a 23-Year-Old Female Patient. J Hematol Clin Res. 2023; 7: 025-028.
DOI: 10.29328/journal.jhcr.1001025
Copyright License: © 2023 Kmira Z, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Acquired factor VII deficiency; Mechanisms; Treatment

A Rare Case of an Acquired Isolated Factor VII Deficiency was Discovered in a 23-Year-Old Female Patient
Zahra Kmira1*, Sassi Nedia1, Ben Yahia Noura1, Ben Sayed Nesrine1, Greisha Ahmed2, Mootameri Wided2, Bouteraa Walid1, Zaier Monia1, Ben Youssef Yosra1, Brahem Nejia2, Haifa Regaieg1 and Khelif Abderrahim1
1Department of Clinical Hematology, Farhat Hached University Hospital, Sousse, Tunisia
2Departement of Cytology, Farhat Hached University Hospital, Sousse, Tunisia
*Address for Correspondence: Zahra Kmira, Department of Clinical Hematology, Farhat Hached University Hospital, Sousse, Tunisia, Email: [email protected]
Introduction: Factor VII (FVII) deficiency, a rare bleeding disorder, can manifest as an autosomal recessive congenital or an acquired coagulopathy. Acquired FVII deficiency, although infrequently reported, presents unique challenges in understanding its mechanisms and identifying underlying causes.
Case presentation: We present a case of acquired FVII deficiency discovered in a 23-year-old female patient with no apparent underlying disease. The patient exhibited spontaneous ecchymosis and gingival hemorrhage, along with low FVII activity and isolated prolongation of prothrombin time. Extensive laboratory investigations excluded liver dysfunction, familial deficiency, vitamin K deficiency, and inhibitory antibodies. Prompt treatment with Fresh Frozen Plasma (FFP) and bypassing agents resulted in a favorable response and resolution of hematomas.
Conclusion: Acquired FVII deficiency was identified with bleeding symptoms in association with prolonged prothrombin time and a low level of FVII activity. In literature, this deficiency has been associated with various conditions such as sepsis, aplastic anemia, stem cell transplantation, and neoplasms, although approximately 14% of cases remain idiopathic. Clinical outcomes remain generally poor, with limited complete remissions reported.
Coagulation factors belong to a family of plasma glycosylated proteins that should be activated for appropriate blood coagulation. Factor VII (FVII) or proconvertin, when bound to tissue factor, initiates the clotting cascade [1]. Factor VII deficiency is a rare disease that could be autosomal recessive congenital coagulopathy or acquired. The mechanisms of acquired FVII deficiency are still unclear, but multiple hypotheses were established. FVII deficiency is associated with clinical bleeding and isolated prolongation of the Prothrombin Time (PT). It may present with hemorrhages of the Central Nervous (CNS) and Gastrointestinal (GIS) systems during the infantile period or may be asymptomatic until adulthood [2]. The severity of bleeding can be unpredictable and is not always linked with the measured levels of FVII [2]. A variety of diseases are responsible for developing acquired FVII deficiency like sepsis, aplastic anemia, Stem Cell Transplant transplantation (SCT), and in association with some neoplasms [3]. In this report, we present a case of acquired FVII deficiency discovered in a 23-year-old female patient and developed with no underlying disease. We also explain the characteristics, the mechanisms, and the possible treatments for this disease.
A 23-year-old female patient, with no medical history, consulted the Emergency Department (ED) for recent onset ecchymosis and gingival hemorrhage appearing spontaneously with no context of trauma. She had no known personal medical history and did not report any previous personal or familial history of bleeding.
The physical examination of the patient showed a 9 x 7 cm ecchymosis on the right forearm, a right knee hemarthrosis, and a 10 x 3 cm hematoma on the right foot (Figure 1). The rest of the examination showed no further abnormalities.
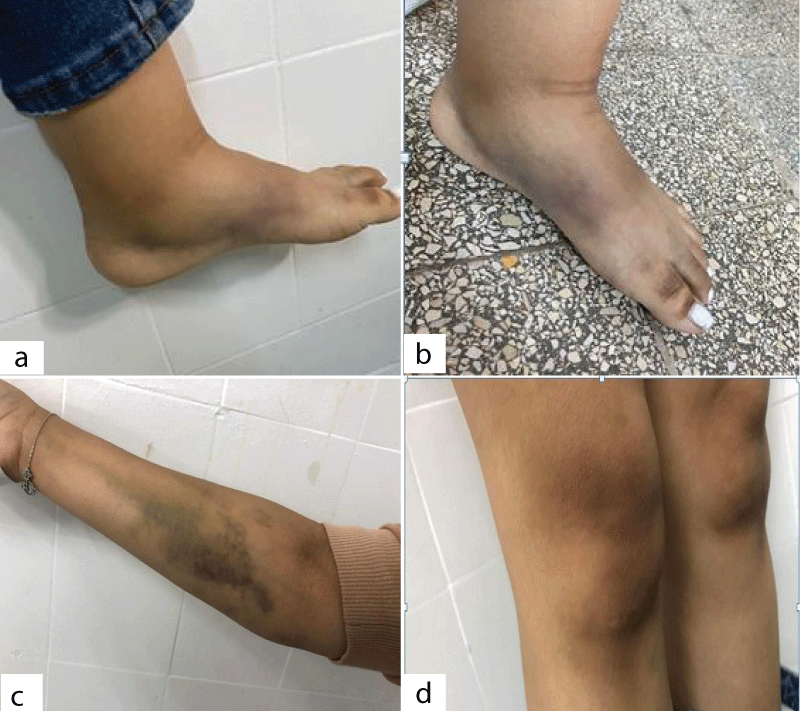
Figure 1: Bleeding symptoms presented in the physical examination of the patient with acquired factor VII deficiency (a and b): A 10 x 3 cm hematoma on the right foot; (c): A 9 x 7 cm ecchymosis on the right forearm; (d): A right knee hemarthrosis.
The Complete Blood Count (CBC) showed a normal white blood cell count at 9 500/mm3, microcytic hypochromic anemia with a level of hemoglobin at 7.4 g/dl, and platelet count at 642 000/mm3. The determination of Prothrombin Time (PT) showed a level of 8,4% (normal>70%) and the Activated Partial Thromboplastin Time (APTT) of 35 secs (normal 25.2 - 35.9 sec). Coagulation screening performed by ACL TOP, a coagulation analyzer, revealed a PT of 13%. A 1:1 mixing study showed correction of PT to 92% immediately and after incubation for 2 hours at 37 Celsius degrees therefore the presence of an inhibitor which may have developed against coagulation factors was ruled out. Also, vitamin K deficiency due to insufficient intake or malabsorption was eliminated since the APTT level was normal, so a FIX, FII, and FX deficiency were unlikely. Factor VII activity level was low at < 5% (normal 50 - 150) and the screening of an inhibitory antibody to FVII was negative, by measuring FVII:C after a cascade of plasma dilution in a diluting factor. Plus, testing for anticoagulant lupus was negative. These findings suggest either the presence of a low titer of inhibitor, which the mixing test will dilute, or the accelerated clearance of FVII due to immune complexes’ formation. The tests were performed using SynthASil.
A summarization of the patient’s characteristics and laboratory findings is represented in Table 1.
| Table 1: A summary of patient's characteristics and laboratory screening. | |
| Patient’s characteristics and laboratory tests | Results |
| Gender | Female |
| Age | 23-year-old |
| Medical history | No |
| Hemoglobin level | 7,4 g/dL |
| Prothrombin Time (PT) | 8,4% (normal>70%) |
| Activated partial thromboplastin time | 35 sec (normal 25.2 - 35.9 sec) |
| PT correction (1:1 ratio) | 92% |
| Factor VII activity level | 1% |
Liver function tests, serum albumin level and plasma electrophoresis were all normal so coagulation factor deficiency secondary to a liver dysfunction was eliminated.
Given our patient’s history, and the clinical and biological findings, the diagnosis of acquired FVII deficiency was confirmed, and no underlying diagnosis was identified. A gene screening was not performed due to its unavailability at the time of diagnosis.
Our patient received a total of 4 Fresh Frozen Plasmas (FFP) units and was put under tranexamic acid, ethamsylate, and I.V. iron therapy with a prompt response to the treatment and gradual resolution of the widespread hematomas.
The patient’s parents and siblings were screened for FVII deficiency, but all the coagulation tests were normal.
Seventeen days later, she presented hemarthrosis of the right elbow. Biology showed the same initial coagulation test abnormalities. The patient was treated with a bypassing agent on account of the function-threatening bleeding.
Factor VII is a glycoprotein that engages in the coagulation process. It binds to the exposed tissue factor after endothelium damage is activated and therefore it activates factor X which is necessary for blood clotting. The protein FVII plays a crucial role in starting the extrinsic coagulation pathway. It works by binding to Tissue Factor (TF) at the site of a blood vessel injury. This binding then leads to the activation of Factor X, which triggers the production of thrombin [3].
Acquired factor VII deficiency is a rare disorder, with only a few cases published in the literature. It is reported to affect more males than females and could be isolated or associated with other coagulation factors deficiency. The occurrence of the disease is reported to be associated with a context of infections, neoplasms, tumors, and immune deficiency [4].
Acquired and isolated FVII deficiency is an extremely rare bleeding disorder, with only a few cases published in the literature, after eliminating the presence of chronic liver disease, familial factor VII deficiency, and vitamin K deficiency. Little is known about this disorder and its mechanisms are still unclear. The exact incidence of this disease is unknown, and it might be underestimated due to its rarity and being uncommonly diagnosed.
Acquired factor VII deficiency can appear in a variety of clinical symptoms ranging from cutaneous ecchymosis to gastrointestinal bleeding or central nervous system hemorrhages. It can also be asymptomatic. Besides, the severity of the bleeding is not correlated to the depth of the FVII activity deficiency. The clinical presentation of the acquired FVII deficiency is variable in bleeding severity and locations ranging from cutaneous ecchymosis to central nervous system hemorrhages [4]. Moreover, unlike most deficiencies in blood coagulation factors, FVII deficiency exhibits a distinct pattern where there is a weak correlation between FVII clotting activity and the likelihood of experiencing bleeding.
The CBC may show an anemia translating the severity of the bleeding and the absence of thrombocytopenia which excludes the probability of a Disseminated Intravascular Coagulation (DIC). The rest of the blood screening shows an isolated low PT and a normal APTT should eliminate a vitamin K deficiency. Other tests are necessary to eliminate an underlying liver disorder like albumin levels. The diagnosis is confirmed with a low level of FVII activity.
In literature, seventy-nine cases have been documented so far [2-11]. The most common causes of acquired FVII deficiency described in the literature were severe systemic sepsis, chronic packed cell transfusion, neoplasia (Solid tumors, acute myeloid leukemia, multiple myeloma…), and stem cell transplantation. It is shown that no underlying disease was correlated to FVII deficiency which may lead to the conclusion that FVII deficiency can be idiopathic in some cases [3,4].
Several hypotheses regarding the exact mechanisms for developing this disease were established, like the excessive consumption of FVII. This is caused by an increase of tissue factor exposure after an alteration of vascular endothelium as found in severe sepsis or intensive chemotherapy. Also, the accelerated catabolism of factor VII was described, resulting from proteases synthesized by leucocytes. Another described mechanism is the presence of a neutralizing antibody directed against FVII that decreases its activity. The presence of the inhibitor can be correlated to the underlying disease thus reappearing with the disease relapse [8,12,14].
In our case, the patient had isolated acquired FVII deficiency, and none of the investigations underlined its cause. The mechanism of acquired FVII deficiency in our patient is unclear suggesting that either the cause preceded the clinical symptoms or that other factors are responsible for the disease development that are yet to be uncovered.
There are no standardized guidelines for the management and treatment of acquired FVII deficiency. The choice of initial treatment must be based on the bleeding severity. FFP is easily available and useful. However, due to the poor half-life of FVII, frequent infusions of FFP are required. Recombinant FVII is necessary to stop the bleeding, especially in life-threatening bleeding, as well as secure hemostasis prior to the invasive procedure which was the case in our patient and in the literature. Whereas no guidelines inform us of the adequate posology nor of the goals to be achieved in terms of biological results [3]. The addition of antifibrinolytic drugs may be helpful to stop the bleeding [15]. Furthermore, finding and treating the underlying disease is necessary to stop the mechanism causing FVII deficiency and to prevent it from recurring again. The use of immunosuppressive agents is recommended in case of the presence of an inhibitor. Treatment with vitamin K is usually not effective [3].
The overall clinical outcome of acquired FVII deficiency described in the literature is generally poor with complete remission seen in only 26 out of 79 patients (33%) [2-5,9,10,12,16,17]. Bleeding complications were the direct cause of death in four patients (intracranial, gastrointestinal, and pulmonary haemorrhage) (5%) [3].
In conclusion, we presented a rare case of isolated acquired FVII deficiency discovered in a 23-year-old female patient without an inhibitor and where no underlying disease was found. Little is known about this disease. The plasma level does not correlate well with the hemorrhagic diathesis. Treatment includes replacement therapy and/or bypassing factors, antifibrinolytics as well as the treatment of underlying disease if exists. Clinical outcome is poor.
Informed consent obtained: Informed consent has been obtained from the patient.
Data availability statement: The dataset of the current study is available from the corresponding author upon motivated request.
- Shahbazi S, Mahdian R. Factor VII Gene Defects: Review of Functional Studies and Their Clinical Implications. Iran Biomed J. 2019 May;23(3):165-74. doi: 10.29252/.23.3.165. Epub 2019 Feb 24. PMID: 30797223; PMCID: PMC6462297.
- Yokuş O, Balçık ÖŞ, Albayrak M. Acquired Factor VII deficiency associated with pneumonia. Journal of Clinical and Experimental Investigations. 2011; 2(4).
- Mulliez SM, Devreese KM. Isolated acquired factor VII deficiency: review of the literature. Acta Clin Belg. 2016 Apr;71(2):63-70. doi: 10.1179/2295333715Y.0000000073. Epub 2016 Mar 3. PMID: 26400474.
- Zaidi MH, Stanley A, Khan M. Acquired Factor VII deficiency - a rare but important consideration. Scott Med J. 2019 Aug;64(3):119-122. doi: 10.1177/0036933019853167. Epub 2019 May 29. PMID: 31142211.
- Abdulsalam MA, Abdulsalam AJ, Özçakar L. Subdural Hematoma From an Acquired Factor VII Deficiency: Is It the Lupus or the Anticoagulant? J Clin Rheumatol. 2021 Dec 1;27(8S):S553-S554. doi: 10.1097/RHU.0000000000001016. PMID: 30896459.
- Abu-Quider A, Asleh M, Fruchtman Y, Ben-Harosh M, Beck G, Abuhasira R, Kapelushnik J. Factor VII Deficiency in Patients Receiving Chronic Packed Cell Transfusions. J Pediatr Hematol Oncol. 2021 Mar 1;43(2):e268-e271. doi: 10.1097/MPH.0000000000001854. PMID: 32520845.
- Anoun S, Lamchahab M, Oukkache B, Qachouh M, Benchekroun S, Quessar A. Acquired factor VII deficiency associated with acute myeloid leukemia. Blood Coagul Fibrinolysis. 2015 Apr;26(3):331-3. doi: 10.1097/MBC.0000000000000024. PMID: 24991944.
- Hammami E, Borgi WE, Lakhal FB, Salem SF, Neji HB, Gouider E. Acquired FVII Deficiency and Acute Myeloid Leukemia: A Case Report and Literature Review. Lab Med. 2022 Sep 1;53(5):e120-e122. doi: 10.1093/labmed/lmab120. PMID: 35181790.
- Matei A, Dolan S, Andrews J, Rivard GÉ. Management of Labour and Delivery in a Patient With Acquired Factor VII Deficiency With Inhibitor: A Case Report. J Obstet Gynaecol Can. 2016 Feb;38(2):160-3. doi: 10.1016/j.jogc.2015.11.002. Epub 2016 Mar 2. PMID: 27032741.
- Ncube M, Majaha K, Nkhori OF, Charumbira T, Mosenye SM, Shailemo D, Rwegerera GM. Acquired Factor VII Deficiency Presenting with Bleeding Diathesis in a 52-Year-Old Black Man in Botswana. Clin Lab. 2022 Mar 1;68(3). doi: 10.7754/Clin.Lab.2021.210702. PMID: 35254019.
- Saif MW, Wasif K, Butler-Bowen H, Miller K, Diasio RB. Acquired factor VII deficiency following FOLFOX in a patient with colorectal cancer who was also DPD deficient. Therap Adv Gastroenterol. 2016 Jan;9(1):121-7. doi: 10.1177/1756283X15604115. PMID: 26770273; PMCID: PMC4699273.
- Anand M, Babbar N. Acquired Factor VII Deficiency in Association with Pyelonephritis. J Assoc Physicians India. 2018 May;66(5):95-6. PMID: 30477072.
- Franchini M, Lippi G, Favaloro EJ. Acquired inhibitors of coagulation factors: part II. Semin Thromb Hemost. 2012 Jul;38(5):447-53. doi: 10.1055/s-0032-1305779. Epub 2012 Jun 27. PMID: 22740184.
- Sevenet PO, Kaczor DA, Depasse F. Factor VII Deficiency: From Basics to Clinical Laboratory Diagnosis and Patient Management. Clin Appl Thromb Hemost. 2017 Oct;23(7):703-710. doi: 10.1177/1076029616670257. Epub 2016 Oct 3. PMID: 27701084.
- Aguilar C, Lucía JF, Hernández P. A case of an inhibitor autoantibody to coagulation factor VII. Haemophilia. 2003 Jan;9(1):119-20. doi: 10.1046/j.1365-2516.2003.00714.x. PMID: 12558789.
- da Silva VA, Silva SS, Martins FF. Acquired deficiency of coagulation factor VII. Rev Bras Hematol Hemoter. 2015 Jul-Aug;37(4):269-71. doi: 10.1016/j.bjhh.2015.05.002. Epub 2015 Jun 3. PMID: 26190433; PMCID: PMC4519705.
- Moosavi L, Bowen J, Coleman J, Heidari A, Cobos E. Acute Myelogenous Leukemia With Trisomy 8 and Concomitant Acquired Factor VII Deficiency. J Investig Med High Impact Case Rep. 2019 Jan-Dec;7:2324709619872657. doi: 10.1177/2324709619872657. PMID: 31496295; PMCID: PMC6734601.